Irina Baranova
Cardiologist
Higher education:
Cardiologist
Kuban State Medical University (KubSMU, KubSMA, KubSMI) Level of education - Specialist 1993-1999
Additional education:
“Cardiology”, “Course on magnetic resonance imaging of the cardiovascular system”,
Research Institute of Cardiology named after. A.L. Myasnikova
"Course on functional diagnostics"
NTsSSKh them. A. N. Bakuleva
"Course in Clinical Pharmacology"
Russian Medical Academy of Postgraduate Education
"Emergency Cardiology"
Cantonal Hospital of Geneva, Geneva (Switzerland)
"Therapy course"
Russian State Medical Institute of Roszdrav
Contacts
Arrhythmogenic right ventricular dysplasia refers to hereditary cardiomyopathies, which are accompanied by structural and functional abnormalities. The main consequence of the pathology is ventricular arrhythmias. Dysplasia causes 11% of sudden cardiac death in people under 30 years of age and 22% of cases among athletes.
Causes of the disease and risk factors
Arrhythmogenic right ventricular cardiomyopathy can be provoked by internal and external factors. Most often, the occurrence of pathology is caused by an aneurysm of the right cardiac ventricle due to an excess of affected tissue in the area of the endocardium and epicardium.
REFERENCE! The disease spreads to people of young (less often) and elderly (more often) age categories. Also, right ventricular dysplasia has a predisposition to hereditary transmission.
People are especially susceptible to the disease if they have the following diseases:
- Congenital pathological or abnormal disorders in the development of the myocardium. This reason for the development of ARVD in most cases leads to accelerated spread of the disease and subsequent death.
- A disturbance in the functioning of metabolism, causing damage to the cardiac tissues of the right ventricle.
- Developing myocarditis.
- Myocardial dysfunction, which is preceded by a decrease in the mass of the heart muscle and its instability in operation.
Also, arrhythmogenic dysplasia occurs in people who are at increased risk of developing the pathology, which is affected by:
- heart rate;
- duration and frequency of arrhythmia;
- excessive acceleration of the heartbeat due to minimal physical activity;
- absence of atrial systole in the presence of atrial fibrillation;
- unsynchronized work of the atrioventricular department;
- the presence of pathological diseases and dysfunctions of the myocardium;
- elderly age;
- the presence of autoimmune and infectious diseases;
- frequent exposure to toxins in the body;
- recent myocardial infarction.
Causes
The etiology of right ventricular arrhythmogenic dysplasia is currently not well understood. Most often, the disease is idiopathic or hereditary in nature. It has been shown that, from a genetic point of view, the cohort of patients is quite heterogeneous; both autosomal dominant and recessive types of inheritance have been identified. In addition, 6 genes and 9 independent loci responsible for the development of right ventricular dysplasia/cardiomyopathy have been identified. Mutant genes associated with right ventricular cardiomyopathy were identified on chromosomes 14 [q23-24] and 17, 12, 18 [q21]. They include intermediate filaments, desmoplakin, plakophilin, plakoglobin, core - factors that protect the myocardium from the effects of mechanical stress at the cellular level. In addition, desmosomes are part of the structure of the intercalary cardiac disc and participate in intracellular signaling networks, which are now being specifically studied in vitro. The manifestation of these mutations is a dysfunction of contractile proteins and their interaction. In addition, there are other variants of PPCM: 1. Congenital anomaly of the development of the pancreatic myocardium with clinical manifestation – sudden death. 2. A consequence of dysplasia caused by metabolic disorders affecting the pancreas and causing progressive replacement of myocytes. 3. Inflammatory origin: dysplasia as a result of myocarditis, when the infection leaves no traces of primary inflammation. According to F. Calabrese et al. , in cases of PPCM myocarditis was often found, and therefore the etiological agent of the disease is considered to be a group of cardiotropic viruses. E. Nurmukhametova considers the most common cause of myocarditis to be damage by the Coxsackie virus group B. In this case, both the conduction system of the heart and the myocardium itself may be involved. But Fontaine takes a different point of view: patients with PPCM are prone to the occurrence of infectious myocarditis, that is, the interpretation of the cause-and-effect relationship is changed. According to Peters, acute/chronic myocarditis leads to involvement of the left side of the heart, which is an unfavorable prognostic sign. Due to conflicting data, the role of infectious myocarditis in PPCM requires further study. 4. According to Turrini, Corrado, PPCM is a consequence of myocardial dystrophy with a decrease in myocardial mass, its dysfunction, electrical instability and heart failure. 5. Morgera et al. Associations of left bundle branch block with arrhythmias and idiopathic ventricular tachyarrhythmias were noted. 6. According to Folino, there is a correlation between a decrease in vagal influence and the severity of the disease.
Forms of the disease
Arrhythmogenic cardiomyopathy, according to the ICD, can manifest itself in several forms. Their classification is as follows:
- The pure form (reference), in which no additional symptoms of the development of the disease are observed.
- Naxos disease, in which cardiomyopathy occurs due to autosomal recessive inheritance (transmitted from older close relatives) and malignant ventricular arrhythmias occur with arrhythmogenic dysplasia of the right ventricle.
- The Venetian form of the disease, the spread of which affects not only the right, but also the left ventricle of the heart muscle. In half of the cases, the disease is inherited and actively manifests itself from the very childhood of the patient, which is why it often ends in death.
- Pokuri disease, which occurs most frequently during adolescence and is fatal, occurs suddenly and is often asymptomatic.
- Tachycardia, manifested in the right ventricle or in heart failure.
- Rare ventricular extrasystoles that occur on the right side of the myocardium and can be fatal.
- Uhl's anomaly, which is the rarest form of manifestation of arrhythmogenic dysplasia. If it is present, the patient rapidly progresses to heart failure with rapid death due to the complete replacement of cardiomyocytes with adipose or fibrous tissue;
- A non-arrhythmogenic form, which often does not have severe symptoms, but can also lead to death.
Causes
The etiology of right ventricular arrhythmogenic dysplasia is currently not well understood. Most often, the disease is idiopathic or hereditary in nature. It has been shown that, from a genetic point of view, the cohort of patients is quite heterogeneous; both autosomal dominant and recessive types of inheritance have been identified.
In addition, 6 genes and 9 independent loci responsible for the development of right ventricular dysplasia/cardiomyopathy have been identified. Mutant genes associated with right ventricular cardiomyopathy were identified on chromosomes 14 [q23-24] and 17, 12, 18 [q21]. They include intermediate filaments, desmoplakin, plakophilin, plakoglobin, core - factors that protect the myocardium from the effects of mechanical stress at the cellular level.
In addition, desmosomes are part of the structure of the intercalary cardiac disc and participate in intracellular signaling networks, which are now being specifically studied in vitro. The manifestation of these mutations is a dysfunction of contractile proteins and their interaction. In addition, there are other variants of PPCM: 1. Congenital anomaly of the development of the pancreatic myocardium with clinical manifestation – sudden death. 2.
A consequence of dysplasia caused by metabolic disorders affecting the pancreas and causing progressive replacement of myocytes. 3. Inflammatory origin: dysplasia as a result of myocarditis, when the infection leaves no traces of primary inflammation. According to F. Calabrese et al. , in cases of PPCM myocarditis was often found, and therefore the etiological agent of the disease is considered to be a group of cardiotropic viruses. E.
Nurmukhametova considers the most common cause of myocarditis to be damage by the Coxsackie virus group B. In this case, both the conduction system of the heart and the myocardium itself may be involved. But Fontaine takes a different point of view: patients with PPCM are prone to the occurrence of infectious myocarditis, that is, the interpretation of the cause-and-effect relationship is changed.
According to Peters, acute/chronic myocarditis leads to involvement of the left side of the heart, which is an unfavorable prognostic sign. Due to conflicting data, the role of infectious myocarditis in PPCM requires further study. 4. According to Turrini, Corrado, PPCM is a consequence of myocardial dystrophy with a decrease in myocardial mass, its dysfunction, electrical instability and heart failure. 5.
Symptoms
The symptoms of all forms of arrhythmogenic cardiac cardiomyopathy (with the exception of nonarrhythmogenic) manifest themselves in the same way, while pronounced signs of pathology are often absent, which complicates the diagnosis of the disease. But there are symptoms that can help determine the presence of the disease:
- tachycardia and its relapses during physical activity of any degree;
- difficulty breathing, shortness of breath due to malfunction of the lungs and decreased vital capacity (VC);
- causeless heart rhythm disturbances;
- pain in the heart area;
- dizziness, causing fainting.
ATTENTION! In cases of complications and exacerbations of the disease, thromboembolism often occurs, which is diagnosed only in a clinical setting.
Symptoms most often appear in adolescence, and especially in males. At the same time, diagnostics often establishes heart failure in such cases, which, with further development, in combination with pathology, causes cardiogenic shock.
In addition, depending on the form of arrhythmogenic dysplasia, individual symptoms of the disease may have different severity. Thus, the asymptomatic (non-arrhythmogenic) form of the pathology rarely has external signs, while the arrhythmic one is characterized by attacks of tachycardia and ventricular extrasystole. The disease, which arises from tachycardia, is manifested by attacks of rapid heartbeat and frequent dizziness due to the development of heart failure.
The duration of symptoms and the period of their manifestation against the background of the development of the disease may differ depending on the patient’s health condition and the diagnosis of the disease.
Epidemiology of ACM
Right ventricular cardiomyopathy is considered a fairly rare disease. It is more often found in patients under forty years of age, mainly in men. According to statistics, every fifth person under the age of 35 who died from a sudden attack of cardiovascular disease had ACM. The disease is also considered one of the leading factors in sudden death among athletes.
Researchers believe that in fact, the prevalence of pathology may be much higher than statistics indicate. After all, the course of the disease is often asymptomatic, and therefore the pathology is not diagnosed. In some countries, the diagnosis of ACM is not made at all, but this does not mean that the disease is truly absent in their territory.
Diagnostics
Diagnosis of arrhythmogenic (dilated) cardiomyopathy is difficult due to mild or absent symptoms, as well as due to the rare occurrence of this disease. An examination of this kind includes exclusively hardware methods, including:
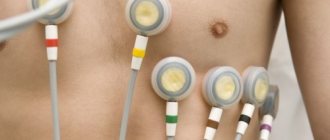
- An ECG, which allows you to find out about the state of the amplitude of the waves, an increase in which can be seen on the electrocardiogram and signals the presence of pathologies of the cardiac region;
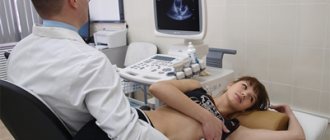
- EchoCG, which determines the size of the heart ventricles and allows them to be compared with normal values;
- MRI, after which it is possible to study the condition of the heart muscle and determine the presence of fatty or fibrinous deposits.
It is also important for the attending physician to pay attention to the family medical history and the medical history of the patient’s closest relatives. Failures in the functioning of the atria and ventricles of the myocardium in them can help diagnose a hereditary disease.
12.4. MYOCARDITIS
Myocarditis is an inflammation of the heart muscle, accompanied by its dysfunction.
The prevalence of myocarditis is unknown, since the disease often occurs in a subclinical form, ending in complete recovery. In men, myocarditis occurs 1.5 times more often than in women.
ETIOLOGY AND PATHOGENESIS
The main causes of myocarditis are listed below.
• Infectious diseases.
• Viruses (Coxsackie, ECHO, adenoviruses, influenza viruses, herpes, cytomegaloviruses, hepatitis B and C, rubella, arboviruses).
• Bacteria (streptococci, staphylococci, borrelia, corynebacterium diphtheria, salmonella, mycobacterium tuberculosis, chlamydia, legionella, rickettsia).
• Protozoa (trypanosomes, toxoplasma).
• Parasites (echinococci, trichinella).
• Fungi (candida, aspergillus, coccidioidomycetes, histoplasma).
• Non-infectious diseases (collagenosis, vasculitis).
• Toxic substances (anthracyclines, catecholamines, cocaine, acetaminophen, lithium).
• Radioactive radiation.
• Allergy (including drug allergies - to penicillins, ampicillin, hydrochlorothiazide, methyldopa, sulfonamides).
In more than 50% of cases, myocarditis is caused by viruses. Experimental models of viral myocarditis (similar to myocarditis in humans) were obtained using Coxsackie B viruses, adenoviruses, and hepatitis C virus. Using molecular diagnostic techniques (PCR, molecular hybridization), persistence of viral infection in the myocardium was shown in a significant proportion of patients. Myocardial damage can occur as a result of direct damage to cardiomyocytes by the agent itself or its toxins (for example, in diphtheria) or be the result of the body's immune response. After exposure to a damaging agent, an inflammatory infiltrate often (but not necessarily) occurs in the myocardium, which consists primarily of lymphocytes, but may also contain neutrophils, eosinophils and macrophages.
Treatment of the disease
REFERENCE! Modern medicine is in search of an effective way to treat arrhythmogenic cardiomyopathy, since to date no universal cure for the fatal disease has been found.
There are general recommendations that help alleviate the course of the pathology, which are aimed at preventing sudden and asymptomatic death of the patient. These treatment methods include:
- Implantation of a cardioverter-defibrillator that performs an antiarrhythmic function. Its installation is recommended for patients with a history of cardiac arrest due to ventricular tachycardia.
- A surgical intervention to help restore normal heart rhythm.
- Drug therapy with the use of drugs Sotalol, Verapamil, Amiodarone, as well as Carvedilol and ACE inhibitors, the action of which is aimed at treating heart failure.
If there are other disturbances in the functioning of the myocardium with manifesting symptoms, the cardiologist and cardiac surgeon may additionally prescribe therapy using symptomatic drugs.
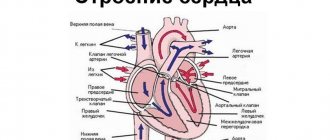
Arrhythmogenic right ventricular cardiomyopathy
Arrhythmogenic right ventricular cardiomyopathy.
*, *, *, #.
* Department of Hospital Therapy No. 2 of the Russian State Medical University (head of the department - corresponding member of the Russian Academy of Medical Sciences, professor), Moscow.
#
Scientific Research Institute of Transplantology and Artificial Organs (director - academician), Moscow .
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is considered a rare disease that has recently received increasing interest. Apparently, there are a certain number of unrecognized cases, while the diagnosis of this disease is often quite possible by fairly simple means.
Under our supervision at the base hospital of our department (MSCh No. 1 AMO ZIL) there were 2 patients with ARVC. A description of one of these cases is presented below.
Patient M, 58 years old, was admitted to the hospital with complaints of shortness of breath at FC level IV. From the anamnesis it is known that she was examined several times for heart disease. It was also known that about 25 years ago the patient suffered from various arrhythmias, including tachycardia with widened QRS complexes.
The patient was hospitalized in the intensive care cardiology department, where the diagnosis of anterior non-Q myocardial infarction was discussed due to the presence of ST segment elevation against the background of incomplete blockade of the right branch of the His bundle (Fig. 1).
X-ray examination (Fig. 2) revealed expansion of the heart shadow in all directions. ECHO-CG revealed significant dilatation of the cavity of the right ventricle (RV) (Fig. 3 A, B), a small amount of fluid in the pericardium (hydropericardium). We excluded congenital heart defects associated with discharge from one chamber of the heart to another. Since the patient had tricuspid regurgitation, the question of the presence of Ebstein's anomaly arose, however, repeated and thorough echocardiographic examination of the tricuspid valve also excluded this pathology.
Get the full text See all projects
Publish articles
During our examination, it was shown that in the anterior wall of the pancreas there is a paradoxical movement (aneurysmal bulge), extending up to the trabecular region.
These data were confirmed by radionuclide ventriculography. Myocardial perfusion scintigraphy synchronized with ECG showed a significant decrease in free wall perfusion, and phase and amplitude images indicated diffuse impairment of RV function. Perfusion of the inferior wall, apical parts of the left ventricle (LV), and interventricular septum (IVS) was also reduced. However, the pattern of RV wall motion, the absence of RV and LV diastolic dysfunction, and the response to a nitroglycerin test excluded the presence of coronary artery stenosis.
The patient was diagnosed with APVC.
Arrhythmogenic right ventricular cardiomyopathy
(traditionally called arrhythmogenic dysplasia of the pancreas) is a disease characterized by the replacement of its myocardium with fibrofatty tissue and clinically manifested by heart failure, ventricular tachycardia with QRS complexes having the morphology of blockade of the left branch of the His bundle and sudden death (SD) (1-4). With a sufficient duration of the disease, regional and then global dysfunction of the RV with its failure occurs, and with further progression of the disease, the LV is affected and biventricular heart failure develops (1-5). The disease is often familial and its etiology is unknown (4).
This condition was first described in 1 The term “dysplasia,” reflecting the dysontogenetic concept of the development of the disease, was used in 1982 (1). However, the disease is now considered not as a developmental anomaly, but as a degenerative process, “dystrophy” or “atrophy” (3). The features of the disease in the definition of the latest classification allowed the International Society and the WHO Federation of Cardiology to include ARVC in the section of cardiomyopathies (12).
Recently, there has been significant progress in understanding the pathogenesis, pathological anatomy and clinic of this condition. However, there is still a lack of information regarding genetics, diagnostic criteria, risk stratification, and the effectiveness of antiarrhythmic therapy in patients with ARVC (4,7).
Pathological anatomy. Morphologically, ARVC manifests itself as diffuse or segmental replacement of the myocardium of the free wall of the pancreas with fibrofatty tissue. The process spreads from the subepicardial layers to the endocardium (1-3).
In 2/3 of cases (3), there are foci of acute myocarditis and round cell inflammatory infiltrates (mainly T-lymphocyte) (8). Due to the lack of clear criteria for normal and pathological fatty infiltration (normally, adipose tissue may be present among myocardiocytes), the disease is often missed in autopsies (3,9).
Two morphological variants of APGC have been described (2):
· fatty form - partial or complete replacement of the pancreatic wall with adipose tissue (the apex or infundibular region is affected) in the absence of fibrosis and inflammatory infiltrates (2-4.9). The LV and interventricular septum are usually not affected, the thickness of the RV wall remains normal. With this option, the risk of VS is controversial (9).
· fibrofatty form – a combination of fatty infiltration with severe fibrosis, inflammatory infiltrates and thinning of the pancreatic wall less than 3 mm (usually its diaphragmatic wall below the posterior cusp of the tricuspid valve) with subsequent formation of aneurysms (1-4). In half of the autopsy cases, aneurysms are located in the inflow, outlet or apical sections of the pancreas (“triangle of dysplasia”) (2,3,27). In this form, the LV and, less commonly, the septum may be involved in the process (2-4.8).
Genetics and epidemiology.
Family history can be traced in 30-50% of cases of ARVC (12,15,28). The most common type of inheritance is autosomal dominant, but an autosomal recessive type is also known. Loci associated with the disease were identified in chromosomes 1, 2, 3 and long arm 14 in the dominant form and in chromosome 17 in the recessive form. The latter is associated with epidermal abnormalities (palmoplantar keratosis - Naxos woolly hair disease). In this condition, the manifestations of the disease are more severe, and penetration in family members reaches 90% (10).
Some familial cases (up to 50%) do not involve these loci, implying genetic heterogeneity.
The prevalence of the disease is unknown. Diagnosis of the disease using routine methods is difficult, so in some cases the disease remains unrecognized until its first manifestation – sudden death (1). A prospective study of cases of VS in young people and athletes was conducted in one of the Italian regions. It has been established that 20% of fatal events in them occur due to previously undiagnosed ARVC (11).
In another proportion of patients, ARVC is not diagnosed until years after its onset, when they develop congestive heart failure (HF) with or without ventricular arrhythmias, often referred to as dilated cardiomyopathy (DCM) (1,3).
In northern Italy, the incidence of ARFA is high, in North America it is low (9). This may be due to the geographical distribution of the disease and insufficient clinical and pathological diagnosis (7).
Etiopathogenesis.
Replacement of the pancreatic myocardium with fibrofatty tissue occurs by three main mechanisms:
1. Apoptosis, or programmed cell death (13). This process is stimulated by cytokines, oxidants, and NO. It is unknown whether apoptosis is triggered by a hereditary abnormality or by an imbalance in the systems that activate and inhibit it.
2. Inflammation, which manifests itself clinically in a variety of ways - acute myocarditis with outcome in fibrosis (3). In severe cases, the LV is also involved (2-4).
3. Myocardial dystrophy, without myocarditis, possibly genetically determined (3).
Clinical diagnosis.
The absence of specific symptoms on physical examination contrasts with the severity of structural changes in the pancreas. Sometimes, as a consequence of pancreatic dysfunction, lengthening and bifurcation of the second tone may be present (1).
Standardized diagnostic criteria were proposed by the ARVC Study Group of the Working Group on Myocardial and Pericardial Diseases of the European Society of Cardiology and the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology (14) (Table 1).
The diagnosis is confirmed if there are 2 major criteria, or 1 major + 2 minor, or 4 minor from different groups. Apparently, the phenotype of ARVC is somewhat broader than is currently understood, so small, borderline normal changes on the ECG or structural abnormalities of the pancreas may also indicate the presence of the disease, especially in families with proven ARVC. Therefore, the presented criteria are specific, but not sensitive enough. In the early stages, the diagnosis of ARVC remains clinical (1,3,4).
To differentiate ARVC from diseases that have similar changes in ECG or cardiac anatomy, the following techniques are used (see Table 2):
· Endomyocardial biopsy is informative regarding fibrofatty replacement of the pancreatic myocardium. The sensitivity of this technique is low, because for safety reasons (risk of perforation of the thinned wall), biopsies are usually taken from the septum, which is rarely involved in the pathological process (16).
· In most cases, the screening method of choice is ECHO-CG.
· Magnetic resonance imaging (MRI) reveals features of the structure and function of the pancreas, especially the presence of adipose tissue in the pancreatic wall (17,18). The sensitivity and specificity of this technique have not been determined, since the quality of the images obtained by different researchers differs, subjectivity remains in the interpretation of the data obtained.
· Electron beam tomography is also a method for verifying the diagnosis; with its help, aneurysmal protrusions and heterogeneity of the pancreatic wall are reliably detected.
· The gold standard for diagnosing ARVC is contrast ventriculography.
· EPI is used to risk stratify patients with ARVC (24) (see Table 3).
Course of the disease.
Electrical instability of the myocardium and progressive ventricular dysfunction determine the natural history of ARVC (1–4,20). Ventricular arrhythmias, from single ventricular extrasystoles (EP) to ventricular tachycardia (VT), sustained or non-sustained, can lead to ventricular fibrillation (VF) and VS (1-4). The imbalance in adrenergic innervation proven in ARVC may play a large role in arrhythmogenesis, since it increases the tendency to VT due to the dispersion of refractory periods and the generation of delayed post-depolarization impulses, usually with physical activity and increasing concentrations of catecholamines (21).
There is long-term clinical and pathological evidence of progression of myocardial damage in ARVC (20). Myocardial contractility decreases, and right ventricular failure occurs. In some patients (up to 76% (24)), the LV is involved in the process, which leads to biventricular HF (2-4,6,20). LV involvement correlates with arrhythmic events, greater severity of cardiomegaly, the presence of inflammatory infiltrates, and HF (34).
The most common causes of death in ARVC are severe ventricular arrhythmia and progressive HF. Mortality rates range from 4 to 20% per year (12,13). The risk of VS is difficult to predict (12) and depends on a history of syncope and complex ventricular arrhythmias (13).
Currently, there is little information about the course of the disease, but it is possible to assume the following clinical phases (19):
1. Latent phase. Structural changes in the RV are minor, but ventricular arrhythmias may occur, which can lead to the first clinical manifestation - sudden death (especially in young people during intense physical activity) (4,11).
2. Obvious electrical disorders. Clinically manifested right ventricular arrhythmias can also lead to ventricular fibrillation. Structural and functional changes in the pancreas become more pronounced (1-5).
3. Global dysfunction of the RV due to progression of the disease and an increase in the volume of myocardial damage, leading to the development of RV failure. LV function is preserved (1,3,20).
Get full text
4. Final stage. Decreased pumping function due to significant LV involvement (3.20). In this case, APZHK can simulate congestive HF due to DCM or other causes. Complications such as atrial fibrillation and thromboembolism occur.
Anomalies of depolarization and repolarization on the ECG.
ECG abnormalities are detected in 75–90% of patients with ARVC (4). The most common is T wave inversion in the precordial leads, which reflect the state of the pancreas (V1-V3). T wave inversion is often associated with slight ST segment elevation (<0.1mV). These repolarization changes are nonspecific and are only minor diagnostic criteria, since they may be a normal variant in women and children under 12 years of age. The same changes can be secondary in BPSV, both isolated and in hereditary heart diseases with overload of the RV.
A wide range of ECG abnormalities reflecting delayed RV excitation includes complete or incomplete SVV, prolongation of the QRS complex in the right precordial leads, and post-excitation waves (epsilon waves - small amplitude potentials following the QRS complex at the beginning of the ST segment). Epsilon waves can be mistaken for the r1 wave in case of incomplete SVPH, as was the case with our patient (see Fig. 1). These ECG changes are more indicative of damage to the myocardium of the interventricular septum (parietal block) than a defect in the conduction system, which is confirmed by precordial mapping data (5).
Prolongation of the QRS complex and epsilon wave are considered major criteria. The duration of the QRS complex more than 110 ms in leads V1-V3 is a relatively sensitive and specific diagnostic marker; the association of QT interval dispersion in 12 leads with the risk of VS has been proven. Epsilon waves are an infrequent phenomenon on a conventional ECG, but they are recorded in more than 30% of patients with ARVC in the form of late potentials on a high-resolution ECG or signal-averaged ECG.
Late potentials are low-amplitude, high-frequency oscillations in the terminal part of the QRS complex. They indicate the presence of areas of slow interventricular conduction, which predisposes to the development of ventricular arrhythmias via the re-entry mechanism. The substrate for this is the islands of surviving myocardiocytes among fibrous and adipose tissue, fragmenting the excitation of the ventricular myocardium. Recently, a relationship has been proven between the presence of late potentials, the volume of pancreatic lesions (the amount of replacement fibrous tissue) and the degree of pancreatic dysfunction (24). There is no connection with re-entry arrhythmias.
A less common ECG abnormality is an increase in the amplitude of P waves of more than 2.5 mV in the right precordial leads.
Symptoms associated with ventricular arrhythmias are palpitations, syncope and presyncope. Ventricular rhythm disturbances in this disease are varied and nonspecific - from single right ventricular ES or persistent VT with the morphology of complexes of the BLVP type to VF leading to VS. The direction of EOS during VT depends on the location of the impulse. In the vertical direction, the EOS is the outflow tract of the pancreas; in the horizontal direction, it is the lower wall. For patients with advanced clinical manifestations of ARVC, VT with different morphology of complexes usually occurs, which suggests the presence of several arrhythmogenic foci in the right ventricle. Therefore, before establishing a diagnosis of ARVC in a patient with right ventricular tachycardia, it is necessary to examine the condition of the right ventricle.
Treatment.
The main problem in ARVC is not the secondary, but the primary prevention of malignant ventricular arrhythmias in patients with asymptomatic disease (24).
Because the clinical features that predict long-term survival in ARVC are poorly understood, it is not known whether beta-adrenergic receptor blockers, other antiarrhythmic drugs, catheter ablation, or an implantable cardioverter defibrillator (ICD) should be used (25). Treatment of patients with ARVC is individualized, the strategy is based on the experience of a particular center. Patients with well-tolerated, non-life-threatening VA typically receive empirical antiarrhythmic therapy with amiodarone, sotalol, beta-blockers, flecainide, propafenone, or a combination thereof (22).
In patients with persistent VT or VF, the selection of antiarrhythmic therapy is guided by the results of EPS against the background of various antiarrhythmics. This may be more effective than empirical selection, even if effectiveness on VS has not been demonstrated. For most patients, sotalol is the drug of choice; its effectiveness in patients with inducible VT is 68%, with non-inducible VT 82% (22), but its ability to prevent VS needs to be confirmed.
Non-drug therapies, such as catheter ablation and ICD therapy, are used in patients with life-threatening ventricular arrhythmias in whom drug therapy is ineffective or has side effects. These methods also have serious side effects. In addition, ventricular arrhythmias induced by EPI may not be reproduced clinically. Despite the superior effect of ICD in preventing VS (23), the exact value of this method needs to be further studied in a larger number of patients.
In patients with progression to severe right-or biventricular HF, treatment consists of conventional therapy, including diuretics, ACE inhibitors, digitalis, and anticoagulants. Such patients are candidates for heart transplantation.
The treatment algorithm for patients with ARVC (24) is presented in Figure 4.
On 01.01.01, the group studying ARVC of the working group on diseases of the myocardium, pericardium and arrhythmias of the European Society of Cardiology, in collaboration with the Scientific Council on Cardiomyopathies of the WHO, created an international registry, the main task of which is to collect data on a large number of patients with a prospective assessment of the accuracy of their diagnosis and long-term survival and effectiveness of therapy (26).
Bibliography
1. Marcus FI, Fontaine G, Guiraudon G, Frank R, Laurenceau JL, Malergue C, Grosgogeat Y. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65:384 –398.
2. Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318:129–133.
3. Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente ML. Arrhythmogenic right ventricular cardiomyopathy: dysplasia, dystrophy, or myocarditis? Circulation. 1996;94:983–991.
4. Nava A, Rossi L, Thiene G, eds. Arrhythmogenic Right Ventricular Cardiomyopathy-Dysplasia. Amsterdam, Netherlands: Elsevier; 1997.
5. Fontaine G, Frank R, Tonet JL, Guiraudon G, Cabrol C, Chomette G, Grosgogeat Y. Arrhythmogenic right ventricular dysplasia: a clinical model for the study of chronic ventricular tachycardia. Jpn Circ J 1984;48:515–538.
Get full text
6. Dalla Volta S, Battaglia G, Zerbini E. Auricularization of right ventricular pressure curve. Am Heart J 1961;61:25–33.
7. Thiene G, Basso C, Danieli GA, Rampazzo A, Corrado D, Nava A. Arrhythmogenic right ventricular cardiomyopathy: a still underrecognized clinic entity. Trends Cardiovasc Med. 1997;7:84–90.
8. Thiene G, Corrado D, Nava A, Rossi L, Poletti A, Boffa GM, Daliento L, Pennelli N. Right ventricular cardiomyopathy: is there evidence of an inflammatory etiology? Eur Heart J 1991;12:22–25.
9. Burke AP, Farb A, Tashko G, Virmani R. Arrhythmogenic right ventricular cardiomyopathy and fatty replacement of the right ventricular myocardium: are they different disease? Circulation. 1998; 97:1571–1580.
10. Coonar AS, Protonotarius N, Tsatsopoulou A, Needham EWA, Houlston RS, Cliff S, Otter MI, Murday VA, Mattu RK, McKenna WJ. Gene for arrhythmogenic right ventricular cardiomyopathy with diffuse nonepidermolytic palmoplantar keratoderma and woolly hair (Naxos disease) maps to 17q21. Circulation. 1998;97:2049–2058.
11. Corrado D, Basso C, Schiavon M, Thiene G. Screening for hypertrophic cardiomyopathy in young athletes. N Engl J Med. 1998;339:364–369.
12. Richardson P, McKenna WJ, Bristow M, Maisch B, Mautner B, O'Connel J, Olsen E, Thiene G, Goodwin J, Gyarfas I, Martin I, Nordet P. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies. Circulation. 1996;93:841–842.
13. Valente M, Calabrese F, Angelini A, Basso C, Thiene G. In vivo evidence of apoptosis in arrhythmogenic right ventricular cardiomyopathy. Am J Pathol. 1998;152:479–484.
14. McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G, Camerini F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Br Heart J 1994;71:215–218.
15. Baraka M, Fontaliran F, Frank R, Fontaine G. Value of task force criteria in the diagnosis of arrhythmogenic right ventricular dysplasia-cardiomyopathy. Herzschr Elektrophys. 1998;9:163–168.
16. Angelini A, Basso C, Nava A, Thiene G. Endomyocardial biopsy in arrhythmogenic right ventricular cardiomyopathy. Am Heart J. 1996; 132:203–206.
17. Blake LM, Schenman MM, Higgins CB. MR features of arrhythmogenic right ventricular dysplasia. Am J Radiol. 1994;162:809–812.
18. Menghetti L, Basso C, Nava A, Angelini A, Thiene G. Spin-echo nuclear magnetic resonance for tissue characterization in arrhythmogenic right ventricular cardiomyopathy. Heart. 1996;76:467–470.
19. Carlson MD, White RD, Throhman RG, Adler LP, Biblio LA, Merkatz KA, Waldo AL. Right ventricular outflow tract ventricular tachycardia: detection of previously unrecognized anatomic abnormalities using cine magnetic resonance imaging. J Am Coll Cardiol. 1994;24:720–727.
20. Pinamonti B, Di Lenarda A, Sinagra G, Silvestri F, Bussani R, Camerini F. Long-term evolution of right ventricular dysplasia-cardiomyopathy. Am Heart J 1995;129:412–415.
21. Wichter T, Hindricks G, Lerch H, Bartenstein P, Borggrefe M, Schober O, Breithardt G. Regional myocardial sympathetic dysinnervation in arrhythmogenic right ventricular cardiomyopathy: an analysis using 123 I-meta-iodobenzylguanidine scintigraphy. Circulation. 1994;89:667–683.
22. Wichter T, Borggrefe M, Hoverkamp W, Chen X, Breithardt G. Efficacy of antiarrhythmic drugs in patients with arrhythmogenic right ventricular disease: results in patients with inducible and noninducible ventricular tachycardia. Circulation. 1992;86:29 –37.
23. Link M, Wang PJ, Haugh CJ, Homoud MK, Foote CB, Costeas XB, Estes NAM III. Arrhythmogenic right ventricular dysplasia: clinical results with implantable cardioverter defibrillator. J Interv Card Electrophysiol. 1997;1:41–48.
Corrado D, Basso C, Thiene G. Arrhythmogenic right ventricular cardiomyopathy: diagnosis, prognosis, and treatment. Heart 2000;83:588–595.
Fontaine G, Zenati O, Tonet J, Hidden F, Himbert C, Frank R. The treatment of ventricular arrhythmias. In: Nava A, Rossi L, Thiene G, eds. Arrhythmogenic Right Ventricular Cardiomyopathy-Dysplasia. Amsterdam, Netherlands: Elsevier; 1997:315–363.
26. Corrado D, Fontaine G, Marcus FI, McKenna WJ, Nava A, Thiene G, Wichter T. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Need for an International Registry. Circulation.2000;101:e101-e106.
27. . Diagnosis and treatment of non-coronary ventricular arrhythmias. Diss. To apply for a step. Ph.D. honey. Sci. Moscow, 1987.
28. . Clinic, diagnosis and treatment of cardiomyopathies. In the book. “Lectures on Cardiology”, ed. And . pp. 14-27, 2001.
Rice. 1. Electrocardiogram of patient M.
Rice. 2. X-ray of the chest organs of patient M.
Rice. 3. EchoCG of patient M. RV dilatation is visible, the left ventricle is intact. The arrow indicates thinning of the pancreatic wall and its aneurysmal protrusion during systole.
A. Apical four-chamber position
B. Parasternal position of the short axis of the LV at the level of the papillary muscles.
Table 1. Criteria for diagnosing ARVC
| Large criteria | Small criteria | ||
| Family history | cases of the disease in several generations of the same family, proven at autopsy or during cardiac surgery | sudden death at a young age (<35 years) in a family member suspected of having ARVD · a single case that meets the criteria for ARVD in the patient’s family | |
| ECG criteria | changes in depolarization and conduction disturbances | epsilon wave or prolongation of QRS complexes in the right chest leads (V1 – V | presence of late potentials on the signal-averaged ECG |
| repolarization changes | T-wave inversion in right precordial leads V2 and V | ||
| Presence of arrhythmias | · persistent or unstable ventricular tachycardia, recorded on an ECG or during SCM or during an exercise test, in which the morphology of the QRS complexes resembles a block of the left branch of the His. · frequent right ventricular extrasystoles (> 1000 in 24 hours) with SCM | ||
| Global or regional dysfunction of the pancreas, detected by echocardiography or angiography, MRI, CRT, radionuclide ventriculography. | · sharp dilatation of the RV and/or decrease in its ejection fraction with or without involvement of the LV in the process multiple or single pancreatic aneurysms, severe segmental dilatation of the pancreas | moderate global dilatation and decreased ejection fraction of the RV with a normal LV moderate segmental dilatation of the pancreas regional dyskinesia of the pancreas | |
| Morphological criteria | replacement of pancreatic myocardial tissue with connective tissue and fat during endomyocardial biopsy |
Table 2. Diseases with which ARVC must be differentiated.
| Diseases | Signs | Diagnostic method | |
| Similar in structure myocardial changes | DCM | Syncope and polymorphic VT are not typical; the process involves mainly the LV, RV damage occurs in the terminal stage of the disease | EchoCG |
| Arrhythmia-induced cardiomyopathy | Syncope is not typical, the LV is primarily affected | EchoCG, medical history | |
| Congenital heart defects · with shunt: ASD, VSD. with damage to the tricuspid valve: Operated tetralogy of Fallot, Ebstein's anomaly, partially anomalous drainage of the pulmonary veins | Characteristic auscultatory pattern | EchoCG, TEE, MRI, cardiac catheterization | |
| Chronic postthromboembolic hypertension | history of pulmonary embolism | Lung scintigraphy, CRT | |
| RV infarction | Clinical picture of a history of myocardial infarction | ECG, EchoCG, radioisotope methods | |
| Uhl Anomaly* | Manifestations of right ventricular HF predominate. In this pathology, the role of apoptosis in the pancreas and conduction system has also been proven (38). | MRI, myocardial biopsy | |
| Similar ECG manifestations | complicated re-entry in the His bundle branch | ||
| Tachycardia due to the re-entry mechanism in pre-excitation syndrome of the Magaym type. | Characteristic ECG signs | EchoCG (absence of characteristic changes in the pancreas), EPI | |
| Sm Brugada* | There are no structural changes in the heart; the most characteristic manifestations (ST segment elevation in the right precordial leads, right bundle branch block, life-threatening rhythm disturbances) are caused by a hereditary defect in the Na channel of myocardiocytes | ECG, EchoCG (no characteristic changes in the pancreas) | |
| Idiopathic tachycardia from the RV outflow tract* | Usually benign and non-familial disease | ECG, EchoCG (no characteristic changes in the pancreas) |
* diseases can be small forms of ARGI (28,37).
Table 3. Goals of EPI in ARVC.
| 1. Identify the possibility of induction of VT or VF during the main protocol or after the use of isoproterenol 2. Determine the hemodynamic significance of persistent VT and the possibility of its degeneration into VF 3. Assess the possibility of interrupting VT using electrical stimulation 4. Possibility of inducing VT during therapy with various antiarrhythmics or implantation of a cardioverter-defibrillator |
Figure 4. Treatment algorithm for patients with ARVC
Possible consequences and complications
The incurable disease arrhythmogenic dysplasia often leads to complications and severe consequences due to the rapid progression of the pathology. These include:
- heart failure and its exacerbation;
- development of thromboembolism;
- cardiac muscle conduction dysfunction;
- aggravation of pathological and chronic arrhythmia;
- pulmonary hypertension;
- sudden cardiac (coronary) death.
ATTENTION! Since it is almost impossible to avoid complications of the disease, it is necessary to monitor your health condition in order to facilitate their course.
control
- For patients with well-tolerated or non-life-threatening ventricular arrhythmias, beta blockers such as sotalol or amiodarone with or without a beta blocker are the most effective drugs with a relatively low proarrhythmic risk.
- For sustained ventricular tachycardia (VT) or ventricular fibrillation (VF), serial therapeutic drug trials using programmed ventricular pacing are used to evaluate efficacy.
- Patients who remain inducible from arrhythmias usually require an ICD, except in rare cases with localized disease where catheter ablation may be an option.
- Radiofrequency ablation results in a significant reduction in VT load in patients with ARVC. [4]
- In patients who are not inducible and present with cardiac arrest or syncope, an automatic ICD is the first option. [5]
- ICDs are the most effective intervention in preventing sudden arrhythmic death. [6]
- The incidence of cardiac and noncardiac mortality after ICD implantation in patients with ARVC is low. [7]
- Treat concomitant heart failure.
- Heart transplant may be required cons of GT;
- ARVC tends to progress with worsening RV function.
- The left ventricle may be involved in the progression of the degenerative process.
- Although AVRC is a progressive disease, the individual triazole [1]
Thank you, we've just sent out a survey email to confirm your preferences.
Based on materials from the site: Patient.info
Prognosis and prevention
The prognosis of a patient’s condition when diagnosed with arrhythmogenic cardiomyopathy can be either favorable (without accelerated development of pathology and its complications) or unfavorable, depending on the following factors:
- Patient's age. Young people are more likely to experience the symptom of sudden death.
- The presence of diseases of this type in the medical history of the patient and immediate relatives.
- Presence of severe tachycardia.
- Cases of syncope.
- Presence of pancreatic dysfunction.
- Presence of chronic heart failure.
IMPORTANT! In the absence of these factors, cardiomyopathy can resolve without a sharp exacerbation of symptoms, with the patient’s stable condition indefinitely.
Therapeutic treatment measures can also reduce the negative consequences of the disease and stop its development.
Preventive measures that overlap with the treatment of dysplasia include the patient’s compliance with the following rules:
- Quitting bad habits: active smoking and frequent alcohol consumption.
- Correction of the diet and regimen, inclusion of healthy foods with vitamin and mineral composition in the menu and minimization of foods high in cholesterol.
- If possible, perform regular minimal physical activity and be active in sports.
These measures also significantly affect the general condition of the patient and have a beneficial effect on treatment.
Forecast
Due to the high degree of variability in the manifestations of pathology, the prognosis is uncertain.
The risk of death during an attack of ventricular fibrillation is critically high, especially if appropriate treatment is not provided. Antiarrhythmic therapy can reduce the patient's risk of death by about a third.
If HF develops, the prognosis is unfavorable. Every tenth patient dies due to thromboembolic complications. The main factor provoking the onset of death is deterioration in the conductivity of muscle fibers and disruptions in heart rhythm.
Prognosis and further screening of patients
Three-year survival rate in patients with DCM is 40%.
The life expectancy of patients with HCM is somewhat longer, but the annual mortality rate still reaches 4% (up to 6% in children). In 10% of patients, spontaneous regression of the disease is observed. The highest mortality rates are in patients with RCMP - mortality over 5 years reaches 70%.
The most favorable outcome is for dishormonal (including in women during menopause) cardiopathy. Subject to adequate hormonal correction of the underlying disease, dystrophic processes regress.
In general, if you follow the recommendations, regularly take medications, timely surgery and correct the underlying disease, the quality of life can be significantly improved.
Patients with CMP should be under medical supervision and undergo examinations once every 2 months. Monitoring of ECG, EchoCG, coagulogram, and INR is mandatory.
There is no specific prevention of the occurrence of CMP. It is very important to conduct a genetic analysis of family members of a patient with established CMP for the purpose of early diagnosis of the disease.
Clinical examination of cardiomyopathy
Your doctor will ask you numerous questions about your symptoms and family history to obtain information to diagnose this disorder.
Often, patients with cardiomyopathy may have close relatives with the disorder or a family history of sudden or premature death. If you have a close relative who was previously well and died suddenly, it may be worth seeing a doctor to investigate the possibility of these disorders.
The doctor will carefully examine the entire cardiovascular system to diagnose these disorders. The nature of the pulse, as well as abnormal and unnecessary heart sounds (tones) are important signs that need to be identified.