Ирина Баранова
Кардиолог
Высшее образование:
Кардиолог
Кубанский государственный медицинский университет (КубГМУ, КубГМА, КубГМИ) Уровень образования — Специалист 1993-1999
Дополнительное образование:
«Кардиология», «Курс по магнитно-резонансной томографии сердечно-сосудистой системы»
НИИ кардиологии им. А.Л. Мясникова
«Курс по функциональной диагностике»
НЦССХ им. А. Н. Бакулева
«Курс по клинической фармакологии»
Российская медицинская академия последипломного образования
«Экстренная кардиология»
Кантональный госпиталь Женевы, Женева (Швейцария)
«Курс по терапии»
Российского государственного медицинского института Росздрава
Контакты
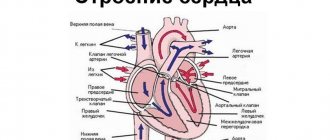
Аритмогенная дисплазия правого желудочка относится к наследственным кардиомиопатиям, которые сопровождаются структурными и функциональными отклонениями. Основное последствие патологии — желудочковые аритмии. Дисплазия приводит к 11% случаев внезапной сердечной смерти у людей до 30 лет и в 22% случаев среди спортсменов.
Причины заболевания и факторы риска
Аритмогенная правожелудочковая кардиомиопатия может быть спровоцирована внутренними и внешними факторами. Наиболее часто возникновение патологии вызывает аневризма правого сердечного желудочка вследствие переизбытка пораженных тканей в области эндокарда и эпикарда.
СПРАВКА! Распространяется заболевание на людей молодой (реже) и пожилой (более часто) возрастных категорий. Также правожелудочковая дисплазия имеет предрасположенность к наследственной передаче.
Особенно часто заболеванию подвержены люди при наличии таких болезней:
- Врожденные патологические или аномальные нарушения в развитии миокарда. Эта причина развития АДПЖ в большинстве случаев ведет к ускоренному распространению заболевания и последующему летальному исходу.
- Нарушение в функционировании метаболизма, провоцирующее поражение сердечных тканей правого желудочка.
- Развивающийся миокардит.
- Дисфункция миокарда, которой предшествует снижение массы сердечной мышцы и ее нестабильность в работе.
Также аритмогенная дисплазия возникает у людей, входящих в группу повышенного риска развития патологии, на что влияет:
- частота сердечного ритма;
- продолжительность и частота возникновения аритмии;
- чрезмерные ускорения сердцебиения вследствие минимальной физической нагрузки;
- отсутствие предсердной систолы при наличии фибрилляции предсердий;
- несинхронизированная работа предсердно-желудочкового отдела;
- наличие патологических заболеваний и дисфункций миокарда;
- пожилой возраст;
- наличие аутоиммунных и инфекционных заболеваний;
- частое воздействие токсинов на организм;
- недавний инфаркт миокарда.
Причины
Этиология правожелудочковой аритмогенной дисплазии в настоящее время изучена недостаточно. Чаще всего заболевание носит идиопатический или наследственный характер. Показано, что с генетической точки зрения когорта больных достаточно гетерогенна, выявлены как аутосомно-доминантные, так и рецессивные типы наследования. Кроме того, идентифицировано 6 генов и 9 независимых локусов, ответственных за развитие правожелудочковой дисплазии/кардиомиопатии. Мутантные гены, ассоциированные с правожелудочковой кардиомиопатией, выявлены в 14 [q23-24] и 17, 12, 18 [q21] хромосомах. Они включают промежуточные филаменты, десмоплакин, плакофиллин, плакоглобин, ядро – факторы защиты миокарда от воздействия механического стресса на клеточном уровне. Помимо этого, десмосомы входят в структуру вставочного сердечного диска и участвуют во внутриклеточных сигнальных сетях, которые сейчас прицельно изучаются in vitro Проявлением данных мутаций является нарушение функции сократительных белков и их взаимодействия. Кроме того, выделяют и другие варианты ПКМП: 1. Врожденная аномалия развития миокарда ПЖ с клиническим проявлением – внезапной смертью. 2. Следствие дисплазии, обусловленной метаболическими нарушениями, поражающими ПЖ и вызывающими прогрессирующее замещение миоцитов. 3. Воспалительного генеза: дисплазия как результат миокардита, когда инфекция не оставляет следов первичного воспаления. По данным F. Calabrese и соавт. , в случаях ПКМП часто обнаруживали миокардит, в связи с чем рассматривают этиологический агент заболевания в виде группы кардиотропных вирусов. Е. Нурмухаметова считает самой частой причиной миокардита поражение вирусом Коксаки группы В. При этом возможно вовлечение как проводящей системы сердца, так и непосредственно миокарда. Но Fontaine придерживается другой точки зрения: пациенты с ПКМП склонны к возникновению инфекционных миокардитов, то есть изменена интерпретация причинно-следственной связи. По мнению Peters, острый/хронический миокардит приводит к вовлечению в процесс левые отделы сердца, что является прогностически неблагоприятным признаком. Ввиду противоречивых данных роль инфекционного миокардита при ПКМП требует дальнейшего изучения. 4. По мнению Turrini, Corrado, ПКМП является следствием дистрофии миокарда с уменьшением массы миокарда, его дисфункцией, электрической нестабильностью и сердечной недостаточностью. 5. Morgera и соавт. Отметили ассоциации блокады левой ножки пучка Гиса с аритмиями и идиопатическими желудочковыми тахиаритмиями. 6. По мнению Folino, существует корреляционная зависимость между снижением вагусного воздействия и степенью тяжести болезни.
Формы болезни
Аритмогенная кардиомиопатия, согласно МКБ, способна проявляться в нескольких формах. Классификация их следующая:
- Чистая форма (эталонная), при которой дополнительной симптоматики развития заболевания не наблюдается.
- Заболевание Наксоса, при котором кардиомиопатия возникает вследствие аутосомно-рецессивного наследования (передается от старших близких родственников) и проявляются злокачественные желудочковые аритмии при аритмогенной дисплазии правого желудочка.
- Венецианская форма болезни, распространение которой поражает не только правый, но и левый желудочек сердечной мышцы. Заболевание в половине случаев передается по наследству и активно проявляет себя с самого детства заболевшего, из-за чего часто заканчивается летальным исходом.
- Заболевание Покури, возникающее наиболее часто в подростковом возрасте и ведущее к смертельным случаям, возникающим внезапно и часто бессимптомно.
- Тахикардия, проявляющаяся в отделе правого желудочка или при заболевании сердечной недостаточностью.
- Редкие экстрасистолы желудочков, которые возникают в правой части миокарда и могут вызвать смертельный исход.
- Аномалия Уля, являющаяся наиболее редкой формой проявления аритмогенной дисплазии. При ее наличии у больного стремительно прогрессирует сердечная недостаточность со скорым летальным исходом вследствие полного замещения кардиомиоцитов жировой или фиброзной тканью;
- Неаритмогенная форма, которая чаще не имеет выраженной симптоматики, но также способна привести к смерти.
Причины
Этиология правожелудочковой аритмогенной дисплазии в настоящее время изучена недостаточно. Чаще всего заболевание носит идиопатический или наследственный характер. Показано, что с генетической точки зрения когорта больных достаточно гетерогенна, выявлены как аутосомно-доминантные, так и рецессивные типы наследования.
Кроме того, идентифицировано 6 генов и 9 независимых локусов, ответственных за развитие правожелудочковой дисплазии/кардиомиопатии. Мутантные гены, ассоциированные с правожелудочковой кардиомиопатией, выявлены в 14 [q23-24] и 17, 12, 18 [q21] хромосомах. Они включают промежуточные филаменты, десмоплакин, плакофиллин, плакоглобин, ядро – факторы защиты миокарда от воздействия механического стресса на клеточном уровне.
Помимо этого, десмосомы входят в структуру вставочного сердечного диска и участвуют во внутриклеточных сигнальных сетях, которые сейчас прицельно изучаются in vitro Проявлением данных мутаций является нарушение функции сократительных белков и их взаимодействия. Кроме того, выделяют и другие варианты ПКМП: 1. Врожденная аномалия развития миокарда ПЖ с клиническим проявлением – внезапной смертью. 2.
Следствие дисплазии, обусловленной метаболическими нарушениями, поражающими ПЖ и вызывающими прогрессирующее замещение миоцитов. 3. Воспалительного генеза: дисплазия как результат миокардита, когда инфекция не оставляет следов первичного воспаления. По данным F. Calabrese и соавт. , в случаях ПКМП часто обнаруживали миокардит, в связи с чем рассматривают этиологический агент заболевания в виде группы кардиотропных вирусов. Е.
Нурмухаметова считает самой частой причиной миокардита поражение вирусом Коксаки группы В. При этом возможно вовлечение как проводящей системы сердца, так и непосредственно миокарда. Но Fontaine придерживается другой точки зрения: пациенты с ПКМП склонны к возникновению инфекционных миокардитов, то есть изменена интерпретация причинно-следственной связи.
По мнению Peters, острый/хронический миокардит приводит к вовлечению в процесс левые отделы сердца, что является прогностически неблагоприятным признаком. Ввиду противоречивых данных роль инфекционного миокардита при ПКМП требует дальнейшего изучения. 4. По мнению Turrini, Corrado, ПКМП является следствием дистрофии миокарда с уменьшением массы миокарда, его дисфункцией, электрической нестабильностью и сердечной недостаточностью. 5.
Симптомы
Симптоматика всех форм аритмогенной кардиомиопатии сердца (за исключением неаритмогенной) проявляется одинаково, при этом ярко выраженные признаки патологии чаще отсутствуют, что усложняет диагностику болезни. Но существуют симптомы, которые способны помочь установить наличие заболевания:
- тахикардия и ее рецидивы при физических нагрузках любой степени;
- трудности с дыханием, одышка из-за сбоев в работе легких и снижения жизненной емкости легких (ЖЕЛ);
- беспричинные сбои сердечного ритма;
- болезненность в области сердца;
- головокружения, провоцирующие обмороки.
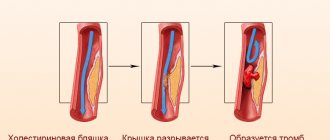
ВНИМАНИЕ! В случаях осложнений и обострений заболевания нередко возникает тромбоэмболия, диагностируемая только в клинических условиях.
Наиболее часто симптоматика проявляется в подростковом возрасте, и особенно у людей мужского пола. При этом диагностика нередко устанавливает в таких случаях сердечную недостаточность, которая при дальнейшем развитии в совокупности с патологией вызывает кардиогенный шок.
Кроме того, в зависимости от формы аритмогенной дисплазии, отдельные симптомы заболевания могут иметь разную выраженность. Так, бессимптомная (неаритмогенная) форма патологии редко имеет внешние признаки, аритмическая — характеризуется приступами тахикардии и экстрасистолии желудочков. Заболевание, возникшее на основе тахикардии, проявляется приступами учащенного сердцебиения и частых головокружений из-за развития сердечной недостаточности.
Продолжительность симптоматики и период ее проявления на фоне развития заболевания могут отличаться в зависимости от состояния здоровья больного и диагностирования болезни.
Эпидемиология АКМП
Правожелудочковая КМП считается достаточно редким заболеванием. Чаще обнаруживается у пациентов до сорока лет, преимущественно у мужчин. Согласно статистике, каждый пятый человек в возрасте до 35 лет, умерший от внезапного приступа сердечно-сосудистого заболевания, был болен АКМП. Также заболевание считается одним из ведущих факторов резкой смерти среди спортсменов.
Исследователи считают, что на самом деле степень распространенности патологии может быть гораздо выше, чем об этом говорит статистика. Ведь течение болезни зачастую бессимптомно, и поэтому патология не диагностируется. В некоторых странах диагноз АКМП вообще не ставят, однако это не означает, что на их территории заболевание действительно отсутствует.
Диагностика
Диагностика аритмогенной (дилатационной) кардиомиопатии вызывает трудности из-за слабого проявления симптоматики или ее отсутствия, а также из-за редкого возникновения данного заболевания. Обследование такого рода включает исключительно аппаратные методы, среди которых:
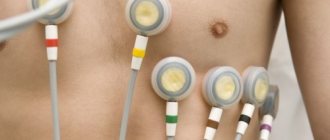
- ЭКГ, позволяющая узнать о состоянии амплитуды зубцов, повышение которой прослеживается на электрокардиограмме и сигнализирует о наличии патологий сердечного отдела;
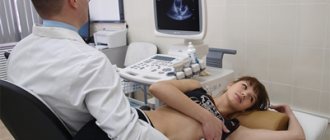
- ЭхоКГ, которая определяет размеры сердечных желудочков и позволяет их сравнить с показателями нормы;
- МРТ, после которой возможно изучить состояние сердечной мышцы и определить наличие жировых или фибринозных отложений.
Также лечащему врачу важно обратить внимание на семейную историю болезни и анамнез ближайших родственников заболевшего. Сбои в работе предсердий и желудочков миокарда у них могут помочь диагностировать заболевание, переданное наследственным путем.
12.4. МИОКАРДИТЫ
Миокардит — воспаление сердечной мышцы, сопровождающееся её дисфункцией.
Распространённость миокардитов неизвестна, поскольку заболевание часто протекает в субклинической форме, заканчиваясь полным выздоровлением. У мужчин миокардит возникает в 1,5 раза чаще, чем у женщин.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Основные причины миокардитов перечислены ниже.
• Инфекционные заболевания.
• Вирусы (Коксаки, ECHO, аденовирусы, вирусы гриппа, герпеса, цитомегаловирусы, гепатита B и C, краснухи, арбовирусы).
• Бактерии (стрептококки, стафилококки, боррелии, коринебактерии дифтерии, сальмонеллы, микобактерии туберкулёза, хламидии, легионеллы, риккетсии).
• Простейшие (трипаносомы, токсоплазмы).
• Паразиты (эхинококки, трихинеллы).
• Грибы (кандиды, аспергиллы, кокцидиоидомицеты, гистоплазмы).
• Неинфекционные заболевания (коллагенозы, васкулиты).
• Токсические вещества (антрациклины, катехоламины, кокаин, ацетаминофен, литий).
• Радиоактивное излучение.
• Аллергия (в том числе лекарственная — на пенициллины, ампициллин, гидрохлоротиазид, метилдопу, сульфаниламиды).
Более чем в 50% случаев миокардиты обусловлены вирусами. Экспериментальные модели вирусных миокардитов (аналогичные миокардиту у человека) получены с использованием вирусов Коксаки B, аденовирусов, вируса гепатита C. С помощью молекулярной диагностической техники (ПЦР, молекулярная гибридизация) показано персистирование вирусной инфекции в миокарде у значительной части больных. Повреждение миокарда может происходить в результате непосредственного повреждения кардиомиоцитов самим агентом или его токсинами (например, при дифтерии) или быть результатом иммунного ответа организма. После воздействия повреждающего агента в миокарде чаще (но не обязательно) возникает воспалительный инфильтрат, который состоит преимущественно из лимфоцитов, но может содержать также нейтрофилы, эозинофилы и макрофаги.
Лечение заболевания
СПРАВКА! Современная медицина пребывает в поисках эффективного способа лечения аритмогенной кардиомиопатии, поскольку на сегодняшний день универсального лекарства от смертельного заболевания найдено не было.
Существуют общие рекомендации, способствующие облегчению течения патологии, которые направлены на предупреждение внезапной и бессимптомной смерти пациента. К таким методам терапии относят:
- Имплантацию кардиовертера-дефибриллятора, выполняющего антиаритмическую функцию. Его установка рекомендована пациентам при наличии в истории болезни остановки сердца вследствие желудочковой тахикардии.
- Хирургическое вмешательство, способствующее восстановлению нормального ритма сердца.
- Медикаментозная терапия с применением препаратов Соталола, Верапамила, Амиодарона, а также Карведилола и ингибиторов АПФ, действие которых направлено на лечение сердечной недостаточности.
При наличии других нарушений в работе миокарда с проявляющейся симптоматикой кардиологом и кардиохирургом может быть дополнительно назначена терапия с применением симптоматических препаратов.
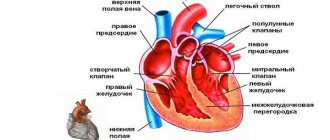
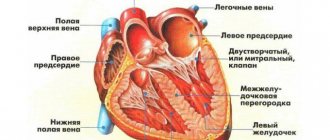
Аритмогенная правожелудочковая кардиомиопатия
Аритмогенная правожелудочковая кардиомиопатия.
*, *, *, #.
* Кафедра госпитальной терапии № 2 РГМУ (зав. кафедрой – член-корр. РАМН, профессор ), Москва.
#
Научно-Исследовательский Институт Трансплантологии и Искусственных Органов (директор – акад. ), Москва.
Аритмогенная правожелудочковая кардиомиопатия (АПЖК) считается редким заболеванием, к которому в последнее время проявляется все больший интерес. По-видимому, имеется определенное количество нераспознанных случаев, в то время, как диагностика этого заболевания часто вполне возможна достаточно простыми средствами.
Под нашим наблюдением на базовой больнице нашей кафедры (МСЧ № 1 АМО ЗИЛа) находились 2 больных с АПЖК. Описание одного их этих случаев преставляется ниже.
Больная М, 58 лет, поступила в стационар с жалобами на одышку на уровне IV ФК. Из анамнеза известно, что она многократно обследована по поводу заболевания сердца. Было также известно, что около 25 лет назад больная страдала от различных аритмий, в том числе, от тахикардии с уширенными QRS-комплексами.
Больная была госпитализирована в реанимационное кардиологическое отделение, где обсуждался диагноз переднего без-Q-инфаркта миокарда из-за наличия элевации сегмента ST на фоне неполной блокады правой ветви пучка Гиса (Рис. 1).
При рентгенологическом исследовании (Рис. 2) выявлено расширение тени сердца во все стороны. При ЭХО-КГ была выявлена значительная дилатация полости правого желудочка (ПЖ) (Рис. 3 А, Б), незначительное количество жидкости в перикарде (гидроперикард). Врожденные пороки сердца, связанные со сбросом из одной камеры сердца в другую, были нами исключены. Поскольку у больной имелась трехстворчатая регургитация, встал вопрос о наличии аномалии Эбштейна, однако многократное и тщательное эхокардиографическое исследование трехстворчатого клапана также исключило эту патологию.
Получить полный текст Смотреть все проекты
Публиковать статьи
В ходе нашего обследования было показано, что в передней стенки ПЖ имеется парадоксальное движение (аневризматическое выбухание), распространяющееся вплоть до трабекулярного отдела.
Эти даные были подтверждены при радионуклидной вентрикулографии. Перфузионная сцинтиграфия миокарда, синхронизированная с ЭКГ, показала значительное снижение перфузии свободной стенки, а фазовые и амплитудные изображения свидетельствовали о диффузном нарушении функции ПЖ. Перфузия нижней стенки, верхушечных отделов левого желудочка (ЛЖ) и межжелудочковой перегородки (МЖП) также оказалась сниженной. Однако, характер движения стенок ПЖ, отстуствие диастолической дисфункции ПЖ и ЛЖ и реакция на пробу с нитроглицерином исключили наличие стенозов коронарных артерий.
Больной был установлен диагноз АПЖК.
Аритмогенная правожелудочковая кардиомиопатия
(традиционно называемая аритмогенной дисплазией ПЖ) – заболевание, характеризующееся замещением его миокарда фиброзно-жировой тканью и проявляющееся клинически сердечной недостаточностью, желудочковой тахикардией с QRS-комплексами, имеющими морфологию блокады левой ветви пучка Гиса и внезапной смертью (ВС) (1-4). При достаточной длительности заболевания возникает регионарная, а затем и глобальная дисфункция ПЖ с его недостаточностью, а при дальнейшем прогрессировании заболевания поражается ЛЖ, и развивается бивентрикулярная сердечная недостаточность (1-5). Заболевание часто семейное, этиология его неизвестна (4).
Впервые это состояние описано в 1Термин “дисплазия”, отражающий дизонтогенетическую концепцию развития заболевания, применен в 1982 г (1). Однако в настоящее время заболевание рассматривают не как аномалию развития, а как дегенеративный процесс, “дистрофию” или “атрофию” (3). Звучащие в определении особенности заболевания при последней классификации позволили Международному Обществу и Федерации Кардиологии ВОЗ включить АПЖК в раздел кардиомиопатий (12).
За последнее время произошел значительный прогресс в понимании патогенеза, патологической анатомии и клиники этого состояния. Однако, до сих пор сохраняется недостаток информации в отношении генетики, диагностических критериев, стратификации риска, эффективности антиаритмической терапии у больных АПЖК (4,7).
Патологическая анатомия. Морфологически АПЖК проявляется диффузным или сегментарным замещением миокарда свободной стенки ПЖ фиброзно-жировой тканью. Процесс распространяется с субэпикардиальных слоев к эндокарду (1-3).
В 2/3 случаев (3) присутствуют очаги острого миокардита и круглоклеточных воспалительных инфильтратов (в основном, Т-лимфоцитарных) (8). Из-за недостаточно четких критериев нормальной и патологической жировой инфильтрации (в норме среди миокардиоцитов может присутствовать жировая ткань) заболевание часто пропускается при аутопсиях (3,9).
Описано два морфологических варианта АПЖК (2):
· жировая форма — частичное или полное замещение стенки ПЖ жировой тканью (поражается верхушка или инфундибулярный отдел) при отстутствии фиброза и воспалительных инфильтратов (2-4,9). ЛЖ, межжелудочковая перегородка обычно не поражаются, толщина стенки ПЖ остается нормальной. При этом варианте риск ВС спорный (9).
· фиброзно-жировая форма – сочетание жировой инфильтрации с выраженным фиброзом, воспалительными инфильтратами и истончением стенки ПЖ менее 3 мм (обычно его диафрагмальной стенки ниже задней створки трикуспидального клапана) с последующим формированием аневризм (1-4). В половине случаев аутопсий аневризмы расположены в приточном, выходном или апикальном отделах ПЖ (“треугольник дисплазии”) (2,3,27). При этой форме в процесс могут вовлекаться ЛЖ и, реже, перегородка (2-4,8).
Генетика и эпидемиология.
Семейный анамнез прослеживается в 30-50% случаев АПЖК (12,15,28). Наиболее частый тип наследования – аутосомно-доминантный, однако известно и об аутосомно-рецессивном типе. Локусы, связанные с заболеванием, выявлены в 1, 2, 3 и длинном плече 14 хромосомах при доминантной форме и в 17 хромосоме при рецессивной. Последняя ассоциирована с эпидермальными аномалиями (пальмоплантарный кератоз — болезнь шерстистых волос острова Наксос). При этом состоянии проявления заболевания более тяжелые, а пенетрация у членов семьи достигает 90% (10).
Некоторые семейные случаи (до 50%) не связаны с поражением этих локусов, что подразумевает генетическую гетерогенность.
Распространенность заболевания не известна. Диагностика болезни рутинными методами сложна, поэтому в части случаев болезнь остается нераспознанной вплоть до первого своего проявления – внезапной смерти (1). В одном из регионов Италии проведено проспективное исследование случаев ВС молодых людей и спортсменов. Установлено, что 20% фатальных событий у них происходит вследствие недиагностированной ранее АПЖК (11).
У другой части больных АПЖК диагностируется лишь спустя годы после начала манифестации, когда у них развивается застойная сердечная недостаточность (СН) с или без желудочковых аритмий, что часто расценивается как дилатационная кардиомиопатия (ДКМП) (1,3).
На севере Италии встречаемость АПЖК высока, в Северной Америке — низка (9). Это может быть связано с географическими особенностями распространения заболевания и недостаточной клинической и патологоанатомической диагностикой (7).
Этиопатогенез.
Замещение миокарда ПЖ фиброзно-жировой тканью происходит по трем основным механизмам:
1. Апоптоз, или программируемая гибель клеток (13). Этот процесс стимулируют цитокины, оксиданты, NO. Неизвестно, запускается ли апоптоз наследственной аномалией или возникновением дисбаланса активирующих и тормозящих его систем.
2. Воспаление, разнообразно проявляющееся клинически – острый миокардит с исходом в фиброз (3). При тяжелом течении вовлекается и ЛЖ (2-4).
3. Дистрофия миокарда, без миокардита, возможно, генетически обусловленная (3).
Клинический диагноз.
Отсутствие специфических симптомов при физикальном осмотре, контрастирует с тяжестью структурных изменений ПЖ. Иногда, как следствие дисфункции ПЖ, может присутствовать удлинение и раздвоенность II тона (1).
Стандартизованные диагностические критерии были предложены Группой по изучению АПЖК Рабочей группы по болезням миокарда и перикарда Европейского Общества Кардиологов и Научного Совета по Кардиомиопатиям Международного Общества и Федерации Кардиологии (14) (Таблица 1).
Диагноз подтверждается при наличии 2 больших критериев, или 1 большого + 2 малых, или 4 малых из разных групп. По-видимому, фенотип АПЖК несколько шире, чем представляется сейчас, поэтому небольшие, пограничные с нормой, изменения на ЭКГ или структурные аномалии ПЖ также могут свидетельствовать о наличии заболевания, особенно в семьях с доказанной АПЖК. Поэтому представленные критерии специфичны, но недостаточно чувствительны. В ранних стадиях диагноз АПЖК остается клиническим (1,3,4).
Для дифференцирования АПЖК от заболеваний, имеющих сходные изменения ЭКГ или анатомии сердца применяются следующие методики (см. таблицу 2):
· Эндомиокардиальная биопсия информативна в отношении фиброзно-жирового замещения миокарда ПЖ. Чувствительность этой методики низка, т. к. по соображениям безопасности (опасность перфорации истонченной стенки) биоптаты обычно берут из перегородки, редко вовлекающейся в патологический процесс (16).
· В большинстве случаев скрининговой методикой выбора является ЭХО-КГ.
· Магнитно-резонансная томография (МРТ) выявляет особенности структуры и функции ПЖ, особенно наличие в стенке ПЖ жировой ткани (17,18). Чувствительность и специфичность этой методики не определена, т. к. качество изображений, получаемых различными исследователями отличается, остается субъективизм при интерпретации полученных данных.
· Электронно-лучевая томография также является методом верификации диагноза, с ее помощью достоверно выявляются аневризматические выпячивания и неоднородность стенки ПЖ.
· Золотым стандартом диагностики АПЖК является контрастная вентрикулография.
· Для стратификации риска пациентов с АПЖК используется ЭФИ (24) (см. таблицу 3).
Течение заболевания.
Электрическая нестабильность миокарда и прогрессирующая дисфункция желудочков определяют естественное течение АПЖК (1-4,20). Желудочковые аритмии, от единичных желудочковых экстрасистол (ЭС) до желудочковой тахикардии (ЖТ), стойкой или нестойкой, могут приводить к фибрилляции желудочков (ФЖ) и ВС (1-4). Доказанный при АПЖК дисбаланс в адренергической иннервации может играть большую роль в аритмогенезе, т. к. при этом увеличивается склонность к ЖТ вследствие дисперсии рефрактерных периодов и генерации отложенных постдеполяризационных импульсов, обычно при физической нагрузке и повышении концентрации катехоламинов (21).
Существуют полученные при долговременном наблюдении клинические и патологоанатомические доказательства прогрессирования поражения миокарда при АПЖК (20). Снижается сократимость миокарда, возникает правожелудочковая недостаточность. У части больных (до 76% (24)) в процесс вовлекается ЛЖ, что приводит к бивентрикулярной СН (2-4,6,20). Поражение ЛЖ коррелирует с аритмическими событиями, большей выраженностью кардиомегалии, наличием воспалительных инфильтратов и СН (34).
Наиболее частые причины смерти при АПЖК – тяжелая желудочковая аритмия и прогрессирующая СН. Смертность колеблется от 4 до 20 % за год (12,13). Риск ВС предсказать сложно (12), он складывается из анамнеза синкопальных состояний и сложных желудочковых аритмий (13).
В настоящее время информации о течении заболевания мало, однако возможно предположить следующие клинические фазы (19):
1. Скрытая фаза. Структурные изменения ПЖ незначительны, однако возможно возникновение желудочковых нарушений ритма, которые могут привести к первому клиническому проявлению – внезапной смерти, (особенно у молодых людей при интенсивной физической нагрузке) (4,11).
2. Явные электрические растройства. Правожелудочковые аритмии, проявляющиеся клинически, также могут привести к фибрилляции желудочков. Структурные и функциональные изменения ПЖ становятся более выраженными (1-5).
3. Глобальная дисфункция ПЖ вследствие прогрессии заболевания и увеличения объема поражения миокарда, приводящая к развитию недостаточности ПЖ. Функция ЛЖ сохранена (1,3,20).
Получить полный текст
4. Финальная стадия. Снижение насосной функции вследствие значительного вовлечения ЛЖ (3,20). При этом АПЖК может симулировать застойную СН вследствие ДКМП или других причин. Возникают такие осложнения как мерцание предсердий и тромбоэмболии.
Аномалии деполяризации и реполяризации на ЭКГ.
У 75-90 % пациентов с АПЖК выявляются аномалии ЭКГ (4). Наиболее частая – инверсия зубца Т в грудных отведениях, отображающих состояние ПЖ (V1-V3). Инверсия зубца Т часто сочетается с небольшой элевацией сегмента ST (< 0,1mV). Эти изменения реполяризации неспецифичны и являются лишь малыми диагностическими критериями, т. к. могут быть вариантом нормы у женщин и детей младше 12 лет. Такие же изменения могут быть вторичными при БПВПГ, как изолированной, так и при наследственных заболеваниях сердца с перегрузкой ПЖ.
Широкий спектр аномалий ЭКГ, отражающий запаздывание возбуждения ПЖ, включает полную или неполную БПВПГ, удлинение комплекса QRS в правых грудных отведениях, волны послевозбуждения (эпсилон-волны — потенциалы небольшой амплитуды, следующие за комплексом QRS в начале сегмента ST). Эпсилон-волны могут быть приняты за зубец r1 при неполной БПВПГ, как это было с нашей больной (см. рис.1). Эти изменения ЭКГ в большей мере свидетельствует о поражении миокарда межжелудочковой перегородки (париетальный блок), чем о дефекте проводящей системы, что подтверждено данными прекордиального картирования (5).
Удлинение QRS-комплекса и эпсилон-волна считаются большими критериями. Продолжительность QRS комплекса более 110 мс в отведениях V1-V3 относительно чувствительный и специфичный диагностический маркер, доказана связь дисперсии интервала QT в 12 отведениях с риском ВС. Эпсилон волны — нечастое явление при обычной ЭКГ, однако они регистрируются более, чем у 30 % больных АПЖК в виде поздних потенциалов при ЭКГ высокого разрешения или сигнал-усредненной ЭКГ.
Поздние потенциалы – низкоамплитудные высокочастотные колебания в конечной части QRS комплекса. Они свидетельствуют о наличии областей замедленного межжелудочкового проведения, что предрасполагает к развитию желудочковых аритмий по механизму re-entry. Субстратом для этого являются островки выживших миокардиоцитов среди фиброзной и жировой ткани, фрагментирующие возбуждение миокарда желудочков. Недавно доказана связь между наличием поздних потенциалов, объемом поражения ПЖ (количеством замещающей фиброзной ткани) и степенью его дисфункции (24). Связи с re-entry-аритмиями нет.
Менее частая ЭКГ-аномалия – увеличение амплитуды зубцов Р более 2,5 мВ в правых грудных отведениях.
Симптомы, связанные с желудочковыми аритмиями – сердцебиение, синкопы и пресинкопальные состояния. Желудочковые нарушения ритма при этом заболевании разнообразны и неспецифичны – от единичных правожелудочковых ЭС или стойкой ЖТ с морфологией комплексов по типу БЛВПГ до ФЖ, ведущей к ВС. Направление ЭОС во время ЖТ зависит от места возникновения импульса. При вертикальном направлении ЭОС это выносящий тракт ПЖ, при горизонтальном направлении – нижняя стенка. Для пациентов с развернутой клиникой АПЖК обычно возникновение ЖТ с разной морфологией комплексов, что предполагает наличие нескольких аритмогенных фокусов в ПЖ. Поэтому, прежде чем установить диагноз АПЖК больному с правожелудочковой тахикардией, необходимо исследовать состояние ПЖ.
Лечение.
Основной проблемой при АПЖК является не вторичная, а первичная профилактика злокачественных желудочковых аритмий у пациентов с бессимптомной формой заболевания (24).
Поскольку клинические проявления, являющиеся предикторами долговременной выживаемости при АПЖК изучены плохо, не известно, по каким критериям выбирать метод лечения — блокаторы бета-адренергических рецепторов, другие антиаритмические препараты, катетерную аблацию или имплантируемый кардиовертер-дефибриллятор (ИКД) (25). Лечение больных с АПЖК индивидуально, стратегия основана на опыте конкретного центра. Больные с хорошо переносимой, нежизнеугрожающей ЖА обычно получают эмпирическую антиаритмическую терапию амиодароном, соталолом, бета-блокаторами, флекаинидом, пропафеноном или их комбинацией (22).
У больных со стойкой ЖТ или ФЖ при подборе антиаритмической терапии руководствуются результатами ЭФИ на фоне различных антиаритмиков. Это может оказаться более эффективным, чем эмпирический подбор, даже если эффективность в отношении ВС не подтверждена. У большинства пациентов препаратом выбора является соталол – его эффективность у больных с индуцируемой ЖТ составляет 68%, с неиндуцируемой 82% (22), однако его способность предотвращать ВС нуждается в подтверждении.
Немедикаментозная терапия, например, катетерная аблация и использование ИКД применяются у больных с жизнеугрожающими желудочковыми аритмиями, у которых лекарственная терапия не эффективна, или проявляются ее побочные действия. Эти методы также имеют серьезные побочные эффекты. Кроме того, индуцируемые при ЭФИ желудочковые аритмии могут не воспроизводиться клинически. Несмотря на наилучший эффект ИКД в отношении профилактики ВС (23), точное значение этого метода нуждается в дополнительном изучении на большем числе пациентов.
У больных с прогрессией в тяжелую право — или бивентрикулярную СН, лечение состоит из обычной терапии, включая диуретики, ИАПФ, препараты дигиталиса, а также антикоагулянты. Такие пациенты — кандидаты на пересадку сердца.
Алгоритм лечения больных АПЖК (24) представлен на рисунке 4.
01.01.01 группой по изучению АПЖК рабочей группы по болезням миокарда, перикарда и аритмиям Европейского Общества Кардиологов в содружестве с Научным советом по Кардиомиопатиям ВОЗ создан международный регистр, основная задача которого – сбор данных о большом числе пациентов с проспективной оценкой точности их диагноза, долговременного выживания и эффективности терапии (26).
Список литературы
1. Marcus FI, Fontaine G, Guiraudon G, Frank R, Laurenceau JL, Malergue C, Grosgogeat Y. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65:384 –398.
2. Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318:129–133.
3. Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente ML. Arrhythmogenic right ventricular cardiomyopathy: dysplasia, dystrophy, or myocarditis? Circulation. 1996;94:983–991.
4. Nava A, Rossi L, Thiene G, eds. Arrhythmogenic Right Ventricular Cardiomyopathy-Dysplasia. Amsterdam, Netherlands: Elsevier; 1997.
5. Fontaine G, Frank R, Tonet JL, Guiraudon G, Cabrol C, Chomette G, Grosgogeat Y. Arrhythmogenic right ventricular dysplasia: a clinical model for the study of chronic ventricular tachycardia. Jpn Circ J. 1984;48:515–538.
Получить полный текст
6. Dalla Volta S, Battaglia G, Zerbini E. Auricularization of right ventricular pressure curve. Am Heart J. 1961;61:25–33.
7. Thiene G, Basso C, Danieli GA, Rampazzo A, Corrado D, Nava A. Arrhythmogenic right ventricular cardiomyopathy: a still underrecognized clinic entity. Trends Cardiovasc Med. 1997;7:84 –90.
8. Thiene G, Corrado D, Nava A, Rossi L, Poletti A, Boffa GM, Daliento L, Pennelli N. Right ventricular cardiomyopathy: is there evidence of an inflammatory etiology? Eur Heart J. 1991;12:22–25.
9. Burke AP, Farb A, Tashko G, Virmani R. Arrhythmogenic right ventricular cardiomyopathy and fatty replacement of the right ventricular myocardium: are they different disease? Circulation. 1998; 97:1571–1580.
10. Coonar AS, Protonotarius N, Tsatsopoulou A, Needham EWA, Houlston RS, Cliff S, Otter MI, Murday VA, Mattu RK, McKenna WJ. Gene for arrhythmogenic right ventricular cardiomyopathy with diffuse nonepidermolytic palmoplantar keratoderma and woolly hair (Naxos disease) maps to 17q21. Circulation. 1998;97:2049–2058.
11. Corrado D, Basso C, Schiavon M, Thiene G. Screening for hypertrophic cardiomyopathy in young athletes. N Engl J Med. 1998;339: 364–369.
12. Richardson P, McKenna WJ, Bristow M, Maisch B, Mautner B, O’Connel J, Olsen E, Thiene G, Goodwin J, Gyarfas I, Martin I, Nordet P. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies. Circulation. 1996;93:841–842.
13. Valente M, Calabrese F, Angelini A, Basso C, Thiene G. In vivo evidence of apoptosis in arrhythmogenic right ventricular cardiomyopathy. Am J Pathol. 1998;152:479–484.
14. McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G, Camerini F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Br Heart J. 1994;71:215–218.
15. Baraka M, Fontaliran F, Frank R, Fontaine G. Value of task force criteria in the diagnosis of arrhythmogenic right ventricular dysplasia-cardiomyopathy. Herzschr Elektrophys. 1998;9:163–168.
16. Angelini A, Basso C, Nava A, Thiene G. Endomyocardial biopsy in arrhythmogenic right ventricular cardiomyopathy. Am Heart J. 1996; 132:203–206.
17. Blake LM, Schenman MM, Higgins CB. MR features of arrhythmogenic right ventricular dysplasia. Am J Radiol. 1994;162:809–812.
18. Menghetti L, Basso C, Nava A, Angelini A, Thiene G. Spin-echo nuclear magnetic resonance for tissue characterization in arrhythmogenic right ventricular cardiomyopathy. Heart. 1996;76:467–470.
19. Carlson MD, White RD, Throhman RG, Adler LP, Biblio LA, Merkatz KA, Waldo AL. Right ventricular outflow tract ventricular tachycardia: detection of previously unrecognized anatomic abnormalities using cine magnetic resonance imaging. J Am Coll Cardiol. 1994;24:720–727.
20. Pinamonti B, Di Lenarda A, Sinagra G, Silvestri F, Bussani R, Camerini F. Long-term evolution of right ventricular dysplasia-cardiomyopathy. Am Heart J. 1995;129:412–415.
21. Wichter T, Hindricks G, Lerch H, Bartenstein P, Borggrefe M, Schober O, Breithardt G. Regional myocardial sympathetic dysinnervation in arrhythmogenic right ventricular cardiomyopathy: an analysis using 123 I-meta-iodobenzylguanidine scintigraphy. Circulation. 1994;89:667–683.
22. Wichter T, Borggrefe M, Hoverkamp W, Chen X, Breithardt G. Efficacy of antiarrhythmic drugs in patients with arrhythmogenic right ventricular disease: results in patients with inducible and noninducible ventricular tachycardia. Circulation. 1992;86:29 –37.
23. Link M, Wang PJ, Haugh CJ, Homoud MK, Foote CB, Costeas XB, Estes NAM III. Arrhythmogenic right ventricular dysplasia: clinical results with implantable cardioverter defibrillator. J Interv Card Electrophysiol. 1997;1:41– 48.
Corrado D, Basso C, Thiene G. Arrhythmogenic right ventricular cardiomyopathy: diagnosis, prognosis, and treatment. Heart 2000;83:588–595.
Fontaine G, Zenati O, Tonet J, Hidden F, Himbert C, Frank R. The treatment of ventricular arrhythmias. In: Nava A, Rossi L, Thiene G, eds. Arrhythmogenic Right Ventricular Cardiomyopathy-Dysplasia. Amsterdam, Netherlands: Elsevier; 1997:315–363.
26. Corrado D, Fontaine G, Marcus FI, McKenna WJ, Nava A, Thiene G, Wichter T. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Need for an International Registry. Circulation.2000;101:e101-e106.
27. . Диагностика и лечение некоронарогенных желудочковых аритмий. Дисс. На соискание степ. канд. мед. наук. Москва, 1987.
28. . Клиника, диагностика и лечение кардиомиопатий. В кн. “Лекции по кардиологии” под ред. и . стр. 14-27, 2001.
Рис. 1. Электрокардиограмма больной М.
Рис. 2. Рентгенография органов грудной полости больной М.
Рис. 3. ЭхоКГ больной М. Видна дилатация ПЖ, левый желудочек интактен. Стрелкой указано истончение стенки ПЖ и его аневризматическое выпячивание в систолу.
А. Апикальная четырехкамерная позиция
Б. Парастернальная позиция короткой оси ЛЖ на уровне папиллярных мышц.
Таблица 1. Критерии диагностики АПЖК
| Большие критерии | Малые критерии | ||
| Семейный анамнез | случаи заболевания в нескольких поколениях одной семьи, доказан-ные на вскрытии или во время кардиохирургической операции | · случай внезапной смерти в молодом возрасте (<35 лет) у члена семьи, у которого подозревалась АДПЖ · единичиный случай, соответствующий критериям АДПЖ в семье больного | |
| ЭКГ критерии | изменения деполяризации и нарушение про-водимости | эпсилон-волна или удлинение QRS-комплексов в праых грудных отведениях (V1 – V | наличие поздних потенциалов на сигнал-усредненной ЭКГ |
| изменения реполяризации | инверсия T-зубцов в правых грудных отведениях V2 и V | ||
| Наличие аритмий | · стойкая или нестойкая желудочковая тахикардия, зафиксированная на ЭКГ или при СХМ или во время исследования с физической нагрузкой, при которой морфология QRS-комплексов напоминает блокаду левой ветви п. Гиса · частые правожелудочковые экстра-систолы (> 1000 за 24 часа) при СХМ | ||
| Глобальная или региональная дисфункция ПЖ, выявляемая при ЭХО-КГ или ангиографии, МРТ, ЭЛТ, радионуклидной вентрикулографии. | · резкая дилатация ПЖ и/или снижение его фракции изгнания с или без вовлечения в процесс ЛЖ · множественные или единичные аневризмы ПЖ, тяжелая сегментарная дилатация ПЖ | · умеренные глобальная дилатация и снижение фракции изгнания ПЖ при нормальном ЛЖ · умеренная сегментарная дилатация ПЖ · региональная дискинезия ПЖ | |
| Морфологические критерии | замещение ткани миокарда ПЖ соединительной тканью и жиром при эндомиокардиальной биопсии |
Таблица 2. Заболевания, с которыми приходится дифференцировать АПЖК.
| Заболевания | Признаки | Диагностический метод | |
| Сходные по структурным изменениям миокарда | ДКМП | Не характерны синкопы, полиморфные ЖТ, в процесс вовлекается преимущественно ЛЖ, поражение ПЖ присоединяется в терминальной стадии заболевания | ЭхоКГ |
| Индуцированная аритмией кардиомиопатия | Не характерны синкопы, первично поражается ЛЖ | ЭхоКГ, анамнез заболевания | |
| Врожденные пороки сердца · с шунтом: ДМПП, ДМЖП. · с поражением трехстворчатого клапана: Оперированная тетрада Фалло, аномалия Эбштейна, частично аномальный дренаж легочных вен | Характерная аускультативная картина | ЭхоКГ, ЧПЭхоКГ, МРТ, катетеризация сердца | |
| Хроническая посттромбоэмболическая гипертензия | ТЭЛА в анамнезе | Сцинтиграфия легких, ЭЛТ | |
| Инфаркт ПЖ | Клиническая картина ИМ в анамнезе | ЭКГ, ЭхоКГ, радиоизотопные методы | |
| Аномалия Уля* | Преобладают проявления правожелудочковой СН. При этой патологии также доказана роль апоптоза в ПЖ и проводящей системе (38). | МРТ, биопсия миокарда | |
| Сходные по ЭКГ-проявлениям | осложненная re-entry в ветви пучка Гиса | ||
| Тахикардия вследствие механизма re-entry при синдроме предвозбуждения по типу с-ма Магайма. | Характерные ЭКГ-признаки | ЭхоКГ (отсутствие характерных изменений ПЖ), ЭФИ | |
| С-м Бругада* | Не возникает структурных изменений сердца, наиболее характерные проявления (элевация сегмента ST в правых грудных отведениях, блокада правой ножки пучка Гиса, жизнеугрожающие нарушения ритма) обусловлены наследственным дефектом Na канала миокардиоцитов | ЭКГ, ЭхоКГ (отсутствие характерных изменений ПЖ) | |
| Идиопатической тахикардией из выходного отдела ПЖ* | Обычно доброкачественное и несемейное заболевание | ЭКГ, ЭхоКГ (отсутствие характерных изменений ПЖ) |
* заболевания могут являтся малыми формами АПЖК (28,37).
Таблица 3. Цели ЭФИ при АПЖК.
| 1. Выявить возможность индукции ЖТ или ФЖ во время основного протокола или после применения изопротеренола 2. Определить гемодинамическую значимость стойкой ЖТ и возможность ее дегенерации в ФЖ 3. Оценить возможность прерывания ЖТ с помощью электростимуляции 4. Возможность индукции ЖТ на фоне терапии различными антиаритмиками или имплантации кардиовертера-дефибриллятора |
Рисунок 4. Алгоритм лечения больных АПЖК
Возможные последствия и осложнения
Неизлечимое заболевание аритмогенной дисплазией часто ведет к проявлению осложнений и тяжелых последствий в связи с быстрым прогрессированием патологии. К ним относятся:
- сердечная недостаточность и ее обострение;
- развитие тромбоэмболии;
- дисфункция проводимости сердечной мышцы;
- отягощение патологической и хронической аритмии;
- легочная гипертензия;
- внезапная сердечная (коронарная) смерть.
ВНИМАНИЕ! Поскольку избежать осложнений заболевания практически невозможно, необходимо контролировать состояние здоровья, чтобы облегчить их протекание.
управление
- Для пациентов с хорошо переносимыми или не угрожающими жизни желудочковыми аритмиями бета-блокаторы, такие как соталол или амиодарон с / без бета-блокатора, являются наиболее эффективными препаратами с относительно низким проаритмическим риском.
- Для устойчивой желудочковой тахикардии (VT) или желудочковой фибрилляции (VF) используются серийные терапевтические испытания лекарственного средства с использованием запрограммированной желудочковой стимуляции для оценки эффективности.
- Пациентам, которые остаются индуцируемыми от аритмий, обычно требуется ICD, за исключением редких случаев с локализованным заболеванием, где катетерная абляция может быть вариантом.
- Радиочастотная абляция приводит к значительному снижению нагрузки на ВТ у пациентов с АРВК. [4]
- У пациентов, которые не являются индуцируемыми и присутствуют при остановке сердца или обмороке, автоматический вариант ICD является первым вариантом. [5]
- ИКД являются наиболее эффективным вмешательством в предотвращении внезапной аритмической смерти. [6]
- Частота сердечной и несердечной смертности после имплантации ICD у пациентов с ARVC низка. [7]
- Лечить сопутствующую сердечную недостаточность.
- Трансплантация сердца может потребоваться минусы GT;
- ARVC имеет тенденцию прогрессировать с ухудшением функции RV.
- Левый желудочек может участвовать в прогрессировании дегенеративного процесса.
- Хотя AVRC является прогрессирующим заболеванием, индивид триазол [1]
Спасибо, мы только что отправили электронное письмо с опросом, чтобы подтвердить ваши предпочтения.
По материалам сайта: Patient.info
Прогноз и профилактика
Прогноз состояния пациента при диагностировании у него аритмогенной кардиомиопатии может быть как благоприятным (без ускоренного развития патологии и ее осложнений), так и неблагоприятным в зависимости от следующих факторов:
- Возраст пациента. Молодые люди чаще подвергаются симптому внезапной смерти.
- Наличие заболеваний подобного типа в анамнезе больного и ближайших родственников.
- Наличие выраженной тахикардии.
- Случаи синкопе.
- Наличие дисфункции поджелудочной железы.
- Наличие хронической сердечной недостаточности.
ВАЖНО! При отсутствии данных факторов кардиомиопатия способна проходить без резкого обострения симптоматики, со стабильным состоянием пациента на неопределенный срок.
Меры терапевтического лечения также способны уменьшить негативные последствия болезни и приостановить ее развитие.
Профилактические меры, перекликающиеся с терапией дисплазии, включают соблюдение больным следующих правил:
- Отказ от вредных привычек: активного курения и частого употребления алкоголя.
- Коррекция режима и рациона питания, включение в меню полезной пищи с витаминно-минеральным составом и минимизация пищи с высоким содержанием холестерина.
- По возможности — выполнение регулярных минимальных физических нагрузок, активные занятия спортом.
Данные мероприятия также существенно влияют на общее состояние пациента и благоприятно сказываются на лечении.
Прогноз
В связи с высокой степенью вариативности проявлений патологии прогноз неопределенный.
Уровень риска смертельного исхода при приступе фибрилляции желудочков критически высок, особенно если не оказывалось соответствующее лечение. Антиаритмическая терапия позволяет снизить угрозу смерти пациента примерно на треть.
В случае формирования СН прогноз неблагоприятный. Каждый десятый больной погибает из-за тромбоэмболических осложнений. Основным фактором, провоцирующим наступление смерти, выступает ухудшение проводимости мышечных волокон и сбои в сердечном ритме.
Прогноз и дальнейший скрининг пациентов
Трехлетняя выживаемость у больных с ДКМП – 40%.
Длительность жизни пациентов с ГКМП несколько дольше, однако годичная смертность все же достигает 4% (до 6% у детей). У 10% пациентов наблюдается спонтанный регресс заболевания. Наиболее высокие показатели летальности у больных РКМП – смертность за 5 лет достигает 70%.
Наиболее благоприятен исход при дисгормональной (в т.ч у женщин в период климакса) кардиопатии. При условии адекватной гормональной коррекции основного заболевания дистрофические процессы регрессируют.
В целом, при соблюдении рекомендаций, регулярном приеме препаратов, своевременном хирургическом вмешательстве и коррекции основного заболевания качество жизни возможно значительно улучшить.
Пациенты с КМП должны находится под диспансерным наблюдением, проходить обследования 1 раз в 2 месяца. Обязателен контроль ЭКГ, ЭхоКГ, коагулограммы, МНО.
Специфической профилактики возникновения КМП не существует. Очень важно проведение генетического анализа членов семьи пациента с установленной КМП с целью ранней диагностики заболевания.
Клиническое обследование кардиомиопатии
Врач задаст вам многочисленные вопросы о симптомах и семейном анамнезе, чтобы получить информацию для диагностики этого расстройства.
Часто пациенты с кардиомиопатией могут иметь близких родственников с расстройством или семейным анамнезом внезапной или преждевременной смерти. Если у вас есть близкий родственник, который ранее чувствовал себя хорошо и внезапно умер, возможно, стоит обратиться к врачу, чтобы выяснить возможность возникновения этих расстройств.
Доктор тщательно изучит всю сердечно-сосудистую систему, чтобы поставить диагноз этих расстройств. Характер пульса, а также аномальные и лишние звуки сердца (тоны) являются важными признаками, которые необходимо выявить.