Vasculitis - what is this disease?
Vasculitis (lat. Vasculum) is the collective name for a group of vascular diseases characterized by inflammation and destruction of the walls of blood vessels - arteries, capillaries, veins and others.
Synonyms for vasculitis are angiitis, arteritis.
By the nature of the pathology, vasculitis resembles atherosclerosis - it is based on thickening of the vessel wall, which reduces the lumen of the bloodstream, disrupts blood circulation, as well as the normal blood supply to one or another part of the body or organ.
Blood, in addition to delivering nutrients to all organs, also delivers oxygen to them. Naturally, due to circulatory disorders, “starving” organs malfunction, and if there is a complete interruption in the supply of blood to them, they begin to die.
The causes of vasculitis are still (as of 2020) not fully understood. There are only assumptions, for example - a combination of genetic features (predisposition), infection (staphylococci, hepatitis viruses) and unfavorable environmental factors.
The classification of vasculitis includes a large number of types and forms, however, depending on the cause, they are divided into primary (an independent disease) and secondary (appears against the background of other diseases). According to localization, there are vasculitis on the skin, in which other organs are not damaged, and internal, the consequences of which can be not only serious cardiovascular diseases, but also death.
By form, the most popular are urticarial, allergic, skin, systemic and hemorrhagic vasculitis.
Vasculitis - ICD
ICD-10: I77.6, I80, L95, M30-M31; ICD-9: 446, 447.6.
Symptoms of the disease
Depending on the classification of AAV, the clinical picture for its different types is heterogeneous, that is, diverse. The symptoms look like this:
- GV (Wegener's granulomatosis). The onset of the pathology is characterized by general weakness and loss of appetite. Later, increased body temperature, myalgia and joint weakness (arthralgia) appear. Further symptoms depend on the affected target organ. If these are the eyes, then the iris and tunica albuginea suffer. If it is the respiratory tract, then first a persistent runny nose with purulent discharge appears. Perforation of the nasal septum is possible. Hemoptysis, ulcerative stomatitis, and purulent discharge from the ears similar to otitis media may appear. If the lungs are affected, the patient experiences shortness of breath, pleurisy, etc. If the target of hemoglobin is the kidneys, then the patient clearly has blood in his urine, a high concentration of protein is noted, and the hemoglobin test is negative. That is, its level is critically reduced.
- Microscopic polyangiitis (MPA). In most cases, the patient exhibits kidney damage in the form of rapidly progressing glomerulonephritis. Symptoms include blood in the urine, increased protein, pain in the lumbar region. If the gastrointestinal tract is affected, stool mixed with blood or vomit with blood cannot be ruled out.
- EGPA. In this case, all the above symptoms are mixed with eosinophilic myo- or endocarditis, pericarditis, and arterial hypertension. Subcutaneous nodules and papules appear on the skin. Over time, ulcers form from the papules. Abdominal pain is noted when the gastrointestinal tract is affected.
Important: the causes of death in AAV are acute renal or heart failure, circulatory arrest, severe internal bleeding, and respiratory failure.
Symptoms of vasculitis
Common symptoms of vasculitis are:
- Increased fatigue, general weakness and malaise;
- Increased body temperature;
- Paleness of the skin;
- Lack of appetite, nausea, sometimes vomiting;
- Loss of body weight;
- Exacerbation of cardiovascular diseases;
- Headaches, dizziness, fainting;
- Visual impairment;
- Sinusitis, sometimes with the formation of nasal polyps;
- Damage to the kidneys, lungs, upper respiratory tract;
- Impaired sensitivity - from minimal to hypersensitivity;
- Arthralgia, myalgia;
- Skin rashes.
The symptoms (clinical manifestations) of vasculitis largely depend on the type, location and form of the disease, so they may differ somewhat, but the main symptom is a violation of normal blood circulation.
Instrumental diagnostics
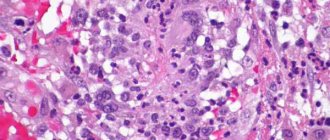
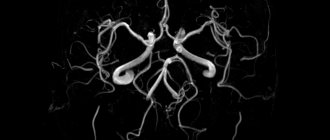
With vasculitis, multiple lesions of the lungs are visible on the fluorogram. An ENT examination reveals chronic sinusitis and otitis media (prolonged inflammation of the paranasal sinuses and inner ear). In biopsy samples of the mucous membranes, granulomatous formations, giant cells and eosinophils are found that infiltrate the tissues. A biopsy of the renal glomeruli determines the presence of ANCA in them. A simpler technique is to measure blood pressure and pulse in both arms. If these indicators are not symmetrical, this is an indirect sign of vascular damage on one side. For certain types of vasculitis, a skin and muscle biopsy is performed. To determine the degree of lung damage and decreased respiratory function, spirography is performed. In order to determine the level of blockage of blood vessels, angiography is performed - an X-ray examination of the vascular bed using contrast agents.
Causes of vasculitis
As we have already mentioned, the etiology of vasculitis is not fully understood, however, there is confirmed data on some of the causes.
Vasculitis can be caused by:
- Genetic predisposition;
- Infection of the body against the background of weakened immunity;
- Overactivity of the immune system to infection;
- Allergic reaction to certain medications;
- Inflammatory processes in the thyroid gland;
- Autoimmune processes;
- Complications of diseases such as reactive arthritis, glomerulonephritis, Schwartz-Jampel syndrome, systemic lupus erythematosus.
Reasons for the development of the disease
All autoimmune diseases, which include Wegener's granulomatosis, appear as a result of disturbances in the functioning of the immune system, when the body reacts aggressively to its own cells, thereby provoking adverse consequences.
There are several reasons for the appearance of this type of vasculitis:
- The disease can become a complication after viral infections. Most often, this diagnosis is given to people who have had infectious mononucleosis, Epstein-Barr virus, influenza, herpes, etc.
- Severe forms of allergic reactions and their improper treatment.
- Excessive interference with the immune system can contribute to the development of the disease. For example, taking drugs to increase the body's resistance can cause an incorrect reaction and lead to irreversible consequences.
- Wegener's granulomatosis can develop after taking strong drugs that are used to treat cancer, tuberculosis and other serious diseases.
- Changes in the hormonal system, such as adolescence, pregnancy or menopause, can also affect a person's immunity.
Unfortunately, the exact cause of the development of Wegener's disease has not yet been identified, so it is worth paying attention to all factors that affect the functioning of the human immune system.
Types of vasculitis
Vasculitis is classified according to the 2012 Chapel Hill Consensus Conference (CHCC) as follows:
By formation:
Primary - the development of the disease is caused by inflammation of the walls of the blood vessels themselves;
Secondary - the development of the disease is due to the reaction of blood vessels against the background of other diseases. Secondary can be:
- Vasculitis associated with hepatitis B virus (HBV);
- Cryoglobulinemic vasculitis associated with hepatitis C virus (HCV);
- Vasculitis associated with syphilis;
- ANCA vasculitis (ANCA) associated with medications;
- Drug-associated immune complex vasculitis;
- Vasculitis associated with cancer (syn. “paraneoplastic vasculitis”)
- Other vasculitis.
By localization:
Vasculitis of large blood vessels:
— Giant cell arteritis (GCA, Horton's disease, temporal arteritis, senile arteritis) is an autoimmune disease characterized by granulomatous inflammation of the main branches of the aorta, most often the branches of the carotid and temporal arteries. In many cases, it is combined with polymyalgia rheumatica, pain and some stiffness in the joints of the pelvic girdle and shoulders, as well as an increase in ESR. The cause is considered to be human infection with hepatitis, herpes, influenza and other viruses. It occurs mainly in people over 50 years of age.
Read also Follicular tonsillitis - symptoms, causes, treatment and prevention
— Takayasu arteritis (nonspecific aortoarteritis) is an autoimmune disease in which a productive inflammatory process develops in the walls of the aorta and its branches, leading them to obliteration. As the disease progresses, pathological processes such as the formation of fibrous granulomas, destruction of elastic fibers, necrotization of smooth muscle cells of the blood vessel wall are observed, after which, after some time, thickening of the intima and medial tunic of the vessel is possible. Sometimes there may be no pulse in the hands, which is why the disease has another name - “pulseless disease.” According to statistics, Takayasu arteritis most often develops in women, in an approximate proportion of men 8 to 1, and patients are young people, from 15 to 30 years old.
Vasculitis of medium-sized vessels:
— Periarteritis nodosa (polyarteritis nodosa, periarteritis nodosa) is an inflammatory disease of the arterial wall of small and medium-sized blood vessels, leading to the development of aneurysms, thrombosis, and heart attacks. At the same time, there is no kidney damage (glomerulonephritis). The main causes are considered to be intolerance to certain medications, as well as persistence of the hepatitis B virus (HBV).
— Kawasaki disease is an acute and febrile disease characterized by inflammatory damage to the walls of small, medium and large-diameter veins and arteries, which is often combined with mucocutaneous lymphatic syndrome. Occurs mainly in children.
Vasculitis of small vessels:
— ANCA-associated vasculitis (AAV):
- Microscopic polyangiitis (MPA) is a not fully understood disease that is associated with the production of antibodies to the cytoplasm of neutrophils, which causes an inflammatory process to develop simultaneously in several organs (most often the lungs and kidneys become victims), and granulomas do not form. Doctors note the following features of the clinical course of GPA: the development of severe pulmonary-renal syndrome (about 50%), damage to the kidneys (about 90%), lungs (from 30 to 70%), skin (about 70%), and visual organs (about 30%). ), peripheral nervous system (about 30%), gastrointestinal tract (about 10%).
- Granulomatosis with polyangiitis (GPA, Wegener's granulomatosis) is a severe and rapidly progressive autoimmune disease characterized by granulomatous inflammation of the walls of small and medium-sized blood vessels (capillaries, venules, arterioles and arteries) involving the eyes, upper respiratory tract, and lungs in the pathological process , kidneys and other organs. If not adequately treated, it can lead to death within 1 year. Doctors note the following features of the clinical course of GPA: damage to the upper respiratory tract (90% or more), kidneys (about 80%), lungs (from 50 to 70%), visual organs (about 50%), skin (from 25 to 35% ), peripheral nervous system (20 to 30%), heart (20% or less), gastrointestinal tract (about 5%).
- Eosinophilic granulomatosis with polyangiitis (EGPA, Churg-Strauss syndrome) is an autoimmune disease caused by an excess of eosinophils in the blood and outside the bloodstream, characterized by granulomatous inflammation of the walls of small and medium-sized vessels involving the upper respiratory tract, lungs, kidneys and other organs in the pathological process. Often accompanied by bronchial asthma, runny nose and other sinusitis, elevated body temperature, shortness of breath, eosinophilia.
— Immune complex vasculitis of small vessels:
- Immunoglobulin-A associated vasculitis (hemorrhagic vasculitis, Henoch-Schönlein purpura, Henoch-Schönlein disease, allergic purpura);
- cryoglobulinemic vasculitis - characterized by damage to the walls of small vessels, mainly the kidneys and skin, the main cause of which is an excessive amount of cryoglobulins in the blood serum, due to which they first settle on the walls of the vessels and then modify them.
- Hypocomplementary urticarial vasculitis (anti-C1q vasculitis);
- Anti-GBM disease.
Vasculitis that can affect blood vessels of varying sizes:
- Behçet's disease is characterized by an inflammatory process in the arteries and veins of small and medium caliber, accompanied by frequent relapses of ulcerative formations on the mucous membranes of the mouth, eyes, skin, genitals, as well as damage to the lungs, kidneys, stomach, brain and other organs.
- Cogan syndrome.
Systemic vasculitis:
- Hemorrhagic vasculitis (Henoch-Schönlein purpura) is characterized by aseptic inflammation of the walls of small vessels (arterioles, venules and capillaries), multiple microthrombosis, developing mainly in the vessels of the skin, kidneys, intestines and other organs. Often accompanied by arthralgia and arthritis. The main cause of hemorrhagic vasculitis is the excessive accumulation of circulating immune complexes in the bloodstream, in which antigens predominate, due to which they settle on the inner surface of the blood wall (endothelium). After re-activation of proteins, the vascular wall changes;
- Lupus vasculitis;
- Behçet's disease;
- Rheumatoid vasculitis;
- Vasculitis in sarcoidosis;
- Takayasu arteritis;
- Other vasculitis.
Read also: Ethmoiditis. Causes, symptoms, types and treatment of ethmoiditis
Vasculitis of individual organs:
- Cutaneous arteritis;
- Cutaneous leukocytclastic angiitis - characterized by an isolated inflammatory process of blood vessels in the skin, without concomitant glomerulonephritis or systemic vasculitis;
- Primary angiitis of the central nervous system;
- Isolated aortritis;
- Other vasculitis.
Systemic vasculitis: modern standards of diagnosis and treatment
Most systemic vasculitis belongs to the ICD-XIII group (systemic connective tissue lesions) and is classified into subgroups M30 and M31. Henoch-Schönlein purpura has the code D69.0, Behcet's disease has the code M35.2, essential cryoglobulinemia has the code D89.1, and thromboangiitis obliterans (Buerger's disease) has the code I73.1. Epidemiology The prevalence of systemic vasculitis ranges from 0.4 to 14 or more cases per 100 thousand population. Systemic vasculitis is a relatively rare disease, but in recent years there has been a tendency to increase its prevalence [1,2,3]. Systemic vasculitis is more common in men than in women and can develop at any age, but mainly in the 4th–5th decades of life, with the exception of hemorrhagic vasculitis and Kawasaki disease, which are often characterized by a predominance of patients in childhood and adolescence. The peak incidence often occurs in winter and spring. Prevention • The etiology of most primary systemic vasculitis is unknown, and therefore primary prevention of these diseases is not carried out. • Some forms of systemic vasculitis (and in individual patients) can be clearly associated with certain causative (or “trigger”) factors, such as hepatitis B virus (classical polyarteritis nodosa), hepatitis C virus (essential cryoglobulinemic vasculitis). In these cases, preventive measures could potentially be aimed at preventing infection of certain risk groups (vaccination against the hepatitis B virus). Classification Depending on the caliber of the affected vessels, the following main forms of systemic vasculitis are distinguished [4]: Damage to large-caliber vessels • Giant cell (temporal) arteritis: granulomatous inflammation of the aorta and its large branches with damage to the extracranial branches of the carotid artery, mainly the temporal artery; usually develops in patients over 50 years of age and is often combined with polymyalgia rheumatica. • Takayasu arteritis: granulomatous inflammation of the aorta and its main branches, usually beginning before the age of 50 years. Damage to medium-sized vessels • Polyarteritis nodosa: necrotizing inflammation of medium and small arteries without glomerulonephritis or vasculitis of arterioles, capillaries and venules. • Kawasaki disease: arteritis, usually found in children, affecting large, medium and small arteries (mainly coronary, sometimes veins) and often combined with mucocutaneous lymphonodular syndrome. Small vessel involvement • Wegener's granulomatosis: granulomatous inflammation involving the respiratory tract and necrotizing vasculitis affecting small and medium-sized vessels (capillaries, venules, arterioles and arteries), with the development of necrotizing glomerulonephritis. • Churg-Strauss syndrome: granulomatous inflammation involving the respiratory tract associated with asthma and eosinophilia, and necrotizing vasculitis affecting small and medium-sized vessels. • Microscopic polyangiitis (polyarteritis): necrotizing vasculitis with a small amount or absence of immune deposits, affecting mainly small vessels (capillaries, venules or arterioles), rarely small and medium-sized arteries, the clinical picture of which is dominated by the phenomena of necrotizing glomerulonephritis and pulmonary capillaritis. • Henoch-Schönlein purpura: vasculitis with predominant IgA deposits, affecting small vessels (capillaries, venules, arterioles), which typically involves the skin, intestines and glomeruli of the kidneys, often combined with arthralgia or arthritis. • Essential cryoglobulinemic vasculitis: vasculitis with cryoglobulin-immune deposits, affecting small vessels (capillaries, venules and arterioles) mainly of the skin and glomeruli of the kidneys and associated with the presence of cryoglobulins in the blood serum. • Cutaneous leukocytoclastic vasculitis: isolated cutaneous leukocytoclastic angiitis without systemic vasculitis or glomerulonephritis. Diagnosis The diagnosis of the disease is based on a detailed clinical examination of the patient and the exclusion of other diseases, and is necessarily confirmed by tissue biopsy data or the results of instrumental and laboratory examination methods. In each case, the disease should be classified according to the classification criteria of the American College of Rheumatology (1990), which, however, are intended for epidemiological studies and not for the diagnosis of systemic vasculitis, especially in the early stages of the disease. Main clinical manifestations • General (constitutional) symptoms: all forms of vasculitis. • Non-destructive oligoarthritis: polyarteritis nodosa, Wegener's granulomatosis, Churg-Strauss syndrome, Henoch-Schönlein purpura. • Skin lesions: – livedo reticularis, digital infarcts, ulcers, nodules: polyarteritis nodosa, Churg-Strauss syndrome, Wegener’s granulomatosis; – palpable purpura: any form of vasculitis, with the exception of giant cell arteritis and Takayasu arteritis. • Multiple mononeuritis: polyarteritis nodosa, cryoglobulinemic vasculitis, Wegener's granulomatosis, Churg-Strauss syndrome. • Kidney damage: – ischemic damage: polyarteritis nodosa, Takayasu arteritis; – glomerulonephritis: microscopic polyangiitis, Wegener’s granulomatosis, cryoglobulinemic vasculitis, Churg-Strauss syndrome, Henoch-Schönlein purpura. • Damage to the ENT organs: Wegener's granulomatosis, less commonly microscopic polyangiitis and Churg-Strauss syndrome. • Lung damage: Wegener's granulomatosis, microscopic polyarteritis, Churg-Strauss syndrome. • Bronchial asthma or allergic rhinitis: Churg-Strauss syndrome. Assessment of activity • When assessing the activity of vasculitis, only signs caused by vasculitis at the time of examination, as well as those that appeared or progressed during the last month before examination of the patient are taken into account [5]. • Depending on the activity, the following phases of the disease are distinguished: – complete remission – absence of signs of activity (total score 0–1 point) and the need for therapy with a normal level of CRP; – partial remission – a decrease in the total score during treatment by 50% of the original; – inactive phase – remission, which does not require maintenance therapy; – “major” exacerbation – involvement of vital organs or systems (lungs, kidneys, central nervous system, cardiovascular system) in the inflammatory process. In this case, there is an increase in the total score by 6 or more points and the prescription of adequate therapy (glucocorticoids and/or cytostatics, intravenous immunoglobulin, plasmapheresis) is required; – “minor” exacerbation – return of the disease with an increase in the total score from 0–1 to 5 points. Recommended inpatient examination Clinical • Assessment of subjective symptoms Laboratory • Complete blood count • Creatinine • Liver enzymes • Creatine phosphokinase: if myopathy is suspected • Complete urinalysis • Bacteriological blood test: exclude infection • Serological tests for syphilis: exclude syphilitic infection • Serological examination: – ANF, RF – exclusion of systemic rheumatic disease; – ANCA – to confirm diagnoses of Wegener’s granulomatosis, microscopic polyangiitis, Churg-Strauss syndrome; – cryoglobulins – to confirm the diagnosis of cryoglobulinemic vasculitis; – APL – exclusion of primary antiphospholipid syndrome; – aBMK – exclusion of Goodpasture syndrome. • Markers of hepatitis B and C virus, HIV (if polyarteritis nodosa and cryoglobulinemic vasculitis are suspected, respectively); cytomegalovirus, Epstein-Barr virus, parvovirus B19 - as needed. Morphological examination An obligatory component of diagnosis (diagnostic criteria) for polyarteritis nodosa, Wegener's granulomatosis, microscopic polyarteritis, Churg-Strauss syndrome, giant cell arteritis. Instrumental examination Angiography: – polyarteritis nodosa – if it is impossible to perform a biopsy or if nonspecific results are obtained; indicated before liver or kidney biopsy to identify microaneurysms that may lead to bleeding during biopsy; – Takayasu arteritis, thromboangiitis obliterans: is important both for confirming the diagnosis and for assessing the dynamics and spread of the inflammatory process. Doppler ultrasound: vascular damage in Takayasu arteritis and thromboangiitis obliterans. R-graphy of the lungs: diagnosis of lung lesions in Wegener's granulomatosis, microscopic polyangiitis, Churg-Strauss syndrome Computer and magnetic resonance imaging: determination of the localization of the process in Wegener's granulomatosis, microscopic polyangiitis, giant cell arteritis, Takayasu arteritis. Differential diagnosis • Clinical signs observed in systemic vasculitis occur in systemic connective tissue diseases (including antiphospholipid syndrome), infections (infective endocarditis, syphilis, other systemic infections) and tumors (atrial myxoma, lymphoproliferative tumors, etc.), severe atherosclerotic lesions vessels. • The need for timely diagnosis of the disease is dictated by the need for early (before the development of irreversible damage to vital organs) administration of aggressive therapy. • Systemic vasculitis should be excluded in all patients with fever, weight loss and signs of multiple organ damage (vascular purpura, multiple mononeuritis, urinary syndrome). Indications for consultation with a rheumatologist: • The presence of any clinical manifestations that suggest systemic vasculitis. Indications for consultation with other specialists: Management of patients with systemic vasculitis requires a multidisciplinary approach. • Dermatologist: vasculitis with skin lesions • Neurologist: when identifying neurological symptoms • Ophthalmologist: for eye damage in patients with Behçet's syndrome, Wegener's granulomatosis, Takayasu's arteritis, giant cell arteritis • Otolaryngologist: for damage to the ENT organs in Wegener's granulomatosis • Nephrologist: kidney damage for systemic necrotizing vasculitis • Pulmonologist: lung damage with Wegener's granulomatosis, microscopic polyarteritis, Churg-Strauss syndrome • Infectious disease specialist and phthisiatrician: carriage of hepatitis B and C viruses, development of opportunistic infections, tuberculosis • Vascular surgeon: vascular damage with Takayasu arteritis • Surgeon: development abdominal pain syndrome with Takayasu arteritis, polyarteritis nodosa, hemorrhagic vasculitis Indications for hospitalization in the rheumatology department: • Clarification of the diagnosis and assessment of the prognosis • Selection of therapy • Exacerbation of the disease • Development of complications Treatment Treatment is always prescribed individually depending on the clinical manifestations of the disease and the type of vasculitis. Goals of therapy • Achieving clinical and laboratory remission • Reducing the risk of exacerbations • Preventing irreversible damage to vital organs • Reducing the risk of developing side effects of drug therapy • Increasing life expectancy. Drug therapy The main groups of drugs used for systemic vasculitis include glucocorticoids and cytostatics (cyclophosphamide, methotrexate, azathioprine) [6,7,8,9,10,11]. For a number of forms of systemic vasculitis, extracorporeal methods of blood purification (plasmapheresis) and the administration of intravenous immunoglobulin are used [8,9,10]. Glucocorticoids • The main treatment for giant cell arteritis and Takayasu arteritis, as well as certain systemic necrotizing vasculitis in the absence of signs of progression, for example, Churg-Strauss syndrome and cryoglobulinemic vasculitis; are used to treat severe forms of hemorrhagic vasculitis with damage to the gastrointestinal tract and kidneys. • Due to the high rate (96%) of disease progression, glucocorticoid monotherapy is not used for the treatment of Wegener's granulomatosis, microscopic polyangiitis and polyarteritis nodosa. • The rapid effect of glucocorticoids can be considered a diagnostic sign of giant cell arteritis and polymyalgia rheumatica. Tactics for prescribing glucocorticoids: • At the onset of the disease, glucocorticoids are usually prescribed in several doses at a dose of 1 mg/kg/day, and then (after 7–10 days) with positive dynamics of clinical and laboratory parameters, they switch to a single dose in the morning. • The duration of suppressive therapy with glucocorticoids is at least 3–4 weeks. • After achieving the effect, the dose of drugs is gradually reduced by 5 mg every 2 weeks. to maintenance (0.15–0.2 mg/kg/day), which is prescribed from 1 year to 3–5 years. • Pulse therapy: – used in patients refractory to standard therapy; – used to induce remission of vasculitis and to suppress its exacerbations (escalation therapy). Cyclophosphamide Drug of choice for: • systemic necrotizing vasculitis (Wegener's granulomatosis, microscopic polyarteritis), polyarteritis nodosa (in the absence of markers of hepatitis B virus replication); • severe forms of hemorrhagic vasculitis and Churg-Strauss syndrome, in which severe, rapidly progressive vascular and renal damage is observed, even despite a good initial clinical response to glucocorticoids. Treatment tactics: • 1–2 mg/kg/day. (orally) for 10–14 days, followed by a decrease depending on the level of leukocytes in the peripheral blood. With very rapid progression of vasculitis, cyclophosphamide is prescribed at a dose of 4 mg/kg/day. per os for 3 days, then 2 mg/kg/day. for 7 days or in the form of pulse therapy (10–15 mg/kg/day) • The total duration of treatment is at least 12 months. after achieving complete remission. Then the dose of the drug is gradually reduced over 2–3 months. 25–50 mg. • Dose selection: the concentration of leukocytes should not be lower than 3000–3500/mm3, and neutrophils – 1000–1500/mm3. At the beginning of treatment, it is advisable to monitor the concentration of leukocytes every other day, and after stabilizing their number - at least once every 2 weeks. • There is evidence of the effectiveness of intermittent therapy with high doses of cyclophosphamide (in combination with glucorticoids) and a decrease in the number of side effects of this drug (500–700 mg/m2 monthly for 6–12 months). However, in Wegener's granulomatosis, this regimen is associated with a high rate of exacerbations [12]. • In patients with renal impairment (serum creatinine > 500 mmol/L), the dose of cyclophosphamide should be reduced by 25–50%. • Treatment with cyclophosphamide should be continued for at least 1 year after remission has been achieved. • Treatment with cyclophosphamide is associated with an increased incidence of side effects (primarily pulmonary infectious complications and bladder cancer in Wegener's granulomatosis), which determine high morbidity and mortality. Azathioprine • Used to maintain remission [13] in necrotizing vasculitis: less likely to cause side effects than cyclophosphamide [14]. • optimal dose – 1–3 mg/kg/day; maintenance dose – 50 mg/day. Methotrexate • Drug dose – 12.5–17.5 mg per week. • In combination with glucocorticoids, it is used to treat Wegener's granulomatosis without rapidly progressive nephritis and severe lung damage, usually in cases of intolerance to cyclophosphamide or to maintain remission of the disease [15,16,17]. • For Takayasu arteritis, the use of methotrexate (17.5 mg/week) in combination with small doses of glucocorticoids allows to achieve remission in 81% of patients and prevent disease progression in 50% of patients, quickly reduce the dose of glucocorticoids and maintain longer remission; low doses of methotrexate (7.5 mg/week) are ineffective [18]. Intravenous immunoglobulin • There are observations of a positive clinical effect when using this drug at a dose of 2 mg/kg (monthly for 6 months) in combination with plasmapheresis for Churg-Strauss syndrome [19]. • Its effectiveness has not been proven in other systemic necrotizing vasculitis. Plasmapheresis • Main indication: as part of combination therapy for acute, progressive disease, manifested by rapidly progressive nephritis (creatinine level more than 500 µmol/l) and severe vasculitis [8,9,10,20]. • In combination with glucocorticoids, they are used for the treatment of essential cryoglobulinemic vasculitis and polyarteritis nodosa associated with the hepatitis B virus. • The use of plasmapheresis in patients with polyarteritis nodosa and Churg-Strauss syndrome, who have unfavorable prognosis factors, does not increase the five-year survival rate of patients compared with standard glucocorticoid therapy and cytostatics [21]; the use of plasmapheresis for the treatment of glomerulonephritis in Churg-Strauss syndrome has no advantages compared to standard therapy for this disease. Combined therapy of systemic necrotizing vasculitis is currently in most cases with systemic necrotic vasculitis (granulomatosis of the vegenera, microscopic polyangita, nodular polyiaritis, Charg -Strass syndrome) is used combined therapy with glucocorticoids and cytostatics [6,8,9,10]. • induction therapy (4-6 months): cyclophosphamide 2 mg/kg/day. Within 1 month. (maximum 150 mg/day); Reduce the dose by 25 mg, if the patient is> 60 years (the number of leukocytes should be> 4.0 × 109/l) in combination with a prenisolone of 1 mg/kg/day. (maximum 80 mg/day); Reduce to 10 mg/day every week. within 6 months. • Supporting therapy: Azatioprine 2 mg/kg/day. or cyclophosphamide 1 mg/kg/day. In combination with a prednisone 5-10 mg/day. • escalation therapy (active severe disease with an increase in creatinine> 500 mmol/l or with pulmonary hemorrhages): 7–10 plasmapheresis procedures within 14 days (plasma removal in the amount of 60 ml/kg with its replacement equal volume of 4.5–5% –In human albumin) or pulse - therapy with methylprednisolone (15 mg/kg/day), dexamethasone (2 mg/kg) for 3 days; If the age of patients <60 years, it is possible to prescribe cyclophosphamide at a dose of 2.5 mg/kg/day. Moopherines of mycophenolat • at a dose of 2 g/day. Used to maintain remission in patients with granulomatosis of the vegener [22,23]. Cyclosporin a • in the initial dose of 5 mg/kg/day. followed by a decrease to 2 mg/kg/day. More effective than colchicine regarding the suppression of progression of uveitis in the disease of Bekhceta [24], and at a dose of 2.5 mg/kg/day. More effective than pulse therapy with cyclophosphamide [25]. • Sometimes it is used to maintain remission in granulomatosis of the vegener with the ineffectiveness of other types of therapy, but its use is limited due to the potential nephrotoxicity of the drug. Leflunomide • There are separate observations about a positive clinical effect when using this drug at a dose of 40 mg/day. In combination with 10 mg/day. prenisolone to maintain remission with granulomatosis of the vegener [26]. Sulfametoxazole/trimetrome • There are reports about the use of sulfametoxazole/trimetrim (160/800 mg 2 times a day) to maintain remission in patients with granulomatosis of the vegener [27], primarily with disabilities (lesion of the ENT -organ) and in the early phase of the disease [ 28]; In a dose (160/800 mg 3 times a week) it is used to prevent infectious complications caused by P.Carinii, against the background of immunosuppressive therapy of methotrexate during the period of maintenance of remission of the disease [29]. • The use of the drug as monotherapy or in combination with prednisone is ineffective to maintain remission with a generalized form of granulomatosis of the vegener. Kolchitsin • In case of illness of behchet, the prescription of colchicin (0.5–1.5 mg/day) reduces the frequency and severity of the exacerbations of the disease and its progression [30]. Antiviral drugs • In the presence of markers of the hepatitis B virus, the prescription of interferon preparations [31] (real -denser, recombinant genetically engineer interferon -a), lamivudina [32] at a dose of 100 mg/day are shown. (lasting up to 6 months) in combination with glucocorticoids and plasmapheresis. • Infection of hepatitis C virus (HCV) for cryoglobulimic vasculitis: Interferon -A, ribavirin preparations in combination with glucocorticoids, cytostatics and plasmapheresis (depending on the severity of the condition) [10, 32]. Pentoxifillin: used to treat systemic vasculitis, especially with vasospastic and ischemic syndromes, skin and kidneys. Surgical treatment • It is indicated in the presence of critical, clinically significant (regional ischemia) stenosis or occlusion of the main arteries for ateritis by Tusas [11], obliterating thrombangiate [33], the development of irreversible (peripheral gangrene) changes in tissue changes; Sublot stenosis for granulomatosis of the vegener (mechanical dilatation of the trachea in combination with the local administration of glucocorticoids) [34]. The prognosis of nodule poly arthol: five -year survival in the background of combined therapy with glucocorticoids and cytostatics is up to 60–80%, the largest death of death is recorded in the first year from the onset of the disease. In most cases, the direct cause of death is cardiovascular disasters. Adverse prognostic factors include: the onset of the disease over the age of more than 50 years, damage to the kidneys, the gastrointestinal tract, central nervous system and the development of cardiomyopathy [35]. Microscopic poly arteritis: the prognosis largely depends on the degree of kidney damage; The five -year survival of patients is 65%, in addition, the cause of death is massive pulmonary bleeding, infectious complications associated with therapy. Vegener's granulomatosis: 5 -year survival is more than 75%; The most common causes of deaths are intercurrent infections, respiratory and renal failure, cardiovascular disasters, malignant neoplasms (bladder cancer). Charga -Strass syndrome: the prognosis of the disease depends on the degree of respiratory failure, the generalization of vasculitis (polyneumnia, glomerulonephritis); 5 -year survival is up to 80%. Purpura Shenlein -Genoch: Despite the fairly frequent recurrence of the disease (in 40% of cases), in general, the prognosis for hemorrhagic vasculitis is favorable. Five -year survival of patients is 95%. During the first two years of disease, complete recovery is observed in 93.9% of children and 89.2% of adults [36]. The main factor that determines the unfavorable prognosis of the disease is a persistent kidney damage. Among all the reasons leading to chronic hemodialysis in pediatric practice, glomerulonephritis with hemorrhagic vasculitis accounts for 3-15% [36]. Gigantic cell arteritis: in general, the forecast for the life of patients is favorable. 5 -year survival of almost 100% [37]. However, there is a serious danger of developing various complications of the disease, first of all, damage to the arteries of the eyes, leading to partial or complete loss of vision. The arteritis by Tsuas: 5-10-15 - survival reaches 80–90%. The most common cause of death is a stroke (50%) and myocardial infarction (about 25%), less often - the rupture of aorta aneurysm (5%). In case of damage to the coronary arteries in the first two years from the moment the symptoms of cardiac pathology occurred, mortality reaches 56%. An unfavorable prognosis is observed in patients, the course of the disease in which is complicated by retinopathy, arterial hypertension, aortic deficiency and aortic aneurysm [38]. In patients with two or more of these syndromes, 10 -year survival from the moment of diagnosis is 58.6%, and most of the fatal outcome falls in the first 5 years of the disease. Crylobulinemic vasculitis: the most common cause of death is the damage to the liver and kidneys, diseases of the cardiovascular system and lymphopoliferative diseases. Literature 1. Koldingsnes W., Nossent H. Epidemiology of Wegener's Granulomatossis in North Norway. Arth.rheum. 2000; 43: 2481–2487. 2. Watts Ra, Lane SE, Bentham G., Scott DGI Epidemiology of Systemic Vasculites: A Ten - Year Study in the United. Arth.rheum. 2000; 43: 414–419. 3. Mahr A., Guillevin L., Poissonnet M., Ayme S. Prevalemces of Polyarterity Nodosa, Microscopic Poliangiitis, Wegener's Granulomatosis and Churg - Strauss Syndrome a French Urban Po Palation in 2000: A Capture - Recapture Estimate. Arth.rheum. 2004; 51: 92–99. 4. Jennette JC, Falk RJ, Andrassy K., et al. Nomenclature of Systemic Vasculitides: Proposed of an International Conference. Arthritis Rheum, 1994; 37: 187–192. 5. Luqmani Ra, Bacon Pa, Moots Rj. et al. Birmingham Vasculites Activity Score (BVAS) In Systemic Necrotising Vasculites.qjmed., 1994; 87: 671–678. 6. Jane D. Evidence - BASED TREATMENT OF SYSTEMIC VASCULITIS. Rheumatology, 2000; 39: 585–595. 7. Ling SM. Polymyalgia Rheumatica and Giant Cell Arteritis. Clinical Guidance from ACP. 2003. 8. Thomas - golbanov C., Sridhan S. Novel Therapies in Vasculites. Expert. Opin. Investig. Drugs. 2001; 10: 1279–1289. 9. Savage Co., Harper L., Ady D. Primary Systemic vasculitis. Lancet, 1997; 349: 553–558. 10. Gross Wl New Concept in Treatment Protocols for Sever Systemic Vasculites. Curr Opin Rheumatol, 1999; 11: 41–46. 11. Sabbadini Mg, Bozzolo E., Baldissera E. Bellone M. Takayasu's Arterites: Therapeutic Strategies. J.Nephrol. 2001; 14: 525–531. 12. Groot KD., ADU D., Savage Co the Value of Pulse Cyclophosphamide in Anca - Sasociated Vasculitis: Meta - Angysis and Critical Review. Nephrol Dial Transplant 2001; 16: 2018–2027. 13. Westman KW, Bygren PG, Olsson H. et al. Relapse Rate, Renal Survival, and Cancer Morbidity in Patience with Wegener's Granulomatosis Or Microscopic Polyangiitis with Renal InvolVeum. J Am Soc Nephrol 1998; 9: 842–852. 14. Jayne Dr, Rasmussen N., Andrassy K. et al. A RANDOMIZED TRIAL of Maintenance Therapy for Vasculites Associated with Antineutrophil Cytoplasmic Autoantibodies New.ingl. J.Med. 2003; 349: 36–44 15. Stone JH, Tun W., Hellman DB Threating of Non - Life Threatening Wegener's Granulomatossis with Methototrexate and Daily Prednisolone as the Initial Therapy of Choice. J.RheumATOL. 1999; 26: 1134–1139. 16. SNELLER MC, Hoffman GS, Talar --williams C. et al. Analysis of 42 Wegener's Granulomatossis Patients Treated with Methotrexate and Prednisone. Arthr.rheum. 1995; 38: 608–613. 17. Langford Ca, Talar --williams C., Barron KS, Sneller MC Use of a Cyclophosphamide - Induction Mithotrexate Regimen for Wegener's Granulomatoissis: Extendence: Extendance D Follow - Up and Rate of Relapse. Am. J. Med. 2003; 114: 463–469. 18. Hoffman GS, Leavitt Ry, Kerr GS et al. Treatment of Glucocorticoid - ReSistant or Relapsing Takayasu Arterity with Methotrexate // Arthr.rheum. - 1994. - Vol. 37. - P.578 - 582. 19. Danieli Mg, Cappelli M., Malcangi G. et al. Long Term Effectivence of Intravenous Immunoglobulin in Churg - Strauss Syndrome. Ann.rheum.dis. 2004; 63: 1649–1654. 20. Cole E., Cattran D., Magil A. et al. A Prospective Randomized Trial of Plasma Exchange as Additive Therapy in Idiopathic Crescentic GlomerulonePhritis. Am J Kidney Dis. 1992; 20: 261–268. 21. Guillevin L., Fain O., Lhote F. et al. Lack of Superiority of Steeroids Plus Plasma Exchange to Steroids Alone in the Treatment of Polyarteritis and Churg - Strauss Syndrome. A proospective, Randomized Trial in 76 Patience // Arthr.rheum. - 1992. - Vol. 35. - P.208 - 215. 22. Nowack R., Gobel U., Klooker P. et al. Mycophenolate Mofetil for Maintenance Therapy of Wegener's Granulomatosis and Microscopic Polyangiitis: A Pilot Study in 11 PATIENTS WITH RENAL INVOLVEMENT. J.AM.SOC. Nephrol. 1999; 10: 1965–1971. 23. Langford Ca, Talar --williams C., Sneller Mc Mycophenolate Mofetil for Remission Maintenance in Therapy of Wegener's Granulomatossis. Athr.rheum. 2004; 51: 278–283. 24. Diaz --llopis M., Cervera M., Menezo Jl Cyclosporin a Treatment of Behcet`s Disease: A Long - Term Study // Curr.eye Res. - 1990. - Vol.9 (SUPPL.). - P.17 - 23. 25. Ozyazgan Y., Yurdakul S., Yazici H. et al. Low Dose Cyclosporin a Versus Pulsed Cyclophosphamide in Behcet's Syndrome: A Single Masked Trial. BR J OPHTHALMOL 1992; 76: 241–247. 26. Metzler C., Low - Friedrich I., Reinold - Keller E. et al. Leflunomide a New Promising Agent in Maintenance of Remission in Wagener's Granulomatosis. Clin.exp.immunol. 1998; 112: 56. 3 Dutch Co - Trimoxazole Wegener Study Group. N.engl.j. Med. 1996; 335: 16–20. 28. Reinhold - Keller E., de Groot K., Rudert H. et al. Response to trimethoprim/Sulfamethoxazole in Wegener's Granulomatossis Depends on the Phase of Disease. QJM 1996; 89: 15–23. 29. Chung JB, Armstrong K., Schwartz JS, Albert D. Cost - Effectivence of Prophylaxis AGainStis Carini Pneumonia in Patents with Wegner's Granulomatosis Undergo Immunosuppressive Therapy. Arthr. Rheum. 2000; 43: 1841–1847. 30. Masuda K., nakajima A., urayama A. et al. Double - Massked Trial of Cyclosporin Versus Colchicne and Long - Term Open Study of Cyclosporin in Behcet`s Disease.lancet. 1989; 1: 1093 - 1096. 31. Guillevin L., Cohen P. Management of Virus --induced Systemic vasculitides. Cur.rheumATOL.Rep. 2002; 4: 60–66. 32. Guillevin L., Mahr A., Cohen P. et al. Short -Term Corticosteroids then lamivudine and Plasma Exchanges to Treat Hepatis b Virus --rerteritis nodosa. Arthr.rheum. 2004; 51: 482–487. 33. TADA Y. Surgical Treatment of Intractable Vasculitis Syndromes with Special Reference to Buerger Disease, Takayasu Arteritis, and So - Called Abdominal Aortic aneurasm. Nippon.rinsho.1994; 52: 2191 - 2202. 34. Hoffman GS, Thomas - Golbanov CK, Chan J. et al. Treatment of Subglottic Stenosis, Due To Wegener's Granulomatosis, With Intraletional Corticosteroids and Dilation. J Rheumatol. 2003; 30: 1017–1025. 35. Guillevin L., Lhote F., Cohen P. et al. CorticoSteroids Plus Cyclophosphamide and Plasma Exchange Versus Corticosteroids Plus Cyclophosphamide Alone in the Treatment of Polyarteritis Nodosa and Churg - Strauss Syndrome iEnts with factors Predicting Poor Prognosis. Arthr.rheum. - 1995. - Vol.38. - P.1638 - 1645. 36. Blanco R., Martinez - Taboada VM, Rodriguez - Valverde V. et al. Henoch - Schonlein Purpura in Adulthood and Childhood. Two Different Expression of the Same Syndrome. Arthr.rheum. 1997; 40: 859 - 864. 37. Salvarani C., Crowson CS, O'Fallon WM et al. Reappraisal of the Epidemiology of Giant Cell Arterites in Olmsted Country, Minnesota, Over a Fifty - Year Period. Arth.rheum. 2004; 51: 264–268. 38. ISHIKAWA K., Maetani S. Long - Term Outcome for 120 Japanese Patients with Takayasu's Disease. Clinical and Statistical Analyses of Related Prognostic Factors // Circulation. - 1994. - Vol.90. - R.855 - 860.
Diagnosis of vasculitis
Diagnosis of vasculitis includes the following examination methods:
- General blood analysis;
- Blood chemistry;
- Ultrasound of the kidneys;
- Echocardiography;
- Electrocardiography (ECG);
- X-ray of the lungs;
- Angiography;
- Biopsy of affected tissues and their further examination.
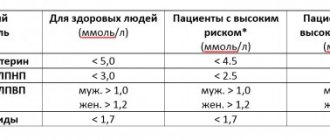
Vasculitis is characterized by an increase in ESR, CRP concentrations, moderate thrombocytosis, normochromic normocytic anemia, antibodies to the cytoplasm of neutrophils (ANCA) and CEC, prolonged sinusitis or otitis, glomerulonephritis.
Laboratory diagnostics
There are basic and additional laboratory methods to determine the diagnosis of autoimmune vascular inflammation. In a general blood test, attention is paid to leukocytosis with an increase in the eosinophil fraction and erythrocyte sedimentation rate. Biochemical testing is also performed to determine an increase in urea and creatinine. Additional tests for vasculitis are prescribed if the disease is already suspected. Therefore, an examination will help confirm the diagnosis. For vasculitis, tests are performed to determine the following indicators:
An ELISA blood test is used to determine ANCA.
- C-reactive protein. This protein is a factor in acute inflammation.
- Alpha and gamma globulins. Their increased content indicates arteritis.
- Antinuclear circulating antibodies (ANCA). Molecules are determined using enzyme-linked immunosorbent assay (ELISA). The ANCA level reflects the activity of the disease.
- Cryoglobulins. Their search is performed for one of the types of vasculitis.
- Red blood cells are intact or destroyed. For this purpose, urine is given.
Return to contents
Treatment of vasculitis
The effectiveness of treatment for vasculitis largely depends on timely and accurate diagnosis, treatment of affected organs and concomitant diseases.
In some cases, the disease goes away on its own, as is the case with primary allergic vasculitis. Complex therapy for vasculitis includes:
1. Drug treatment; 2. Physiotherapeutic methods of treatment; 3. Diet; 4. Preventive measures (at the end of the article).
Important! Before using medications, be sure to consult your doctor!
Drug treatment of vasculitis
Drug treatment of systemic vasculitis is aimed at the following goals:
- Suppression of immunopathological reactions that are the basis of the disease;
- Maintaining stable and long-term remission;
- Treatment of disease relapses;
- Preventing the development of secondary diseases and complications;
Medicines for vasculitis:
Glucocorticoids are a group of hormonal drugs that have anti-inflammatory, antiallergic, immunoregulatory, anti-stress, anti-shock and other properties. In this case, these hormones play one of the most important roles in the treatment of giant cell arteritis (GCA) and Takayasu arteritis, contributing in many cases to achieving stable and long-term remission. In the case of a very rapid response to glucocorticoids, the response can be considered as an additional diagnostic feature of GCA and polymyalgia rheumatica (PPR).
Among the glucocorticoids we can distinguish: Prednisolone, Hydrocortisone.
Cytostatic drugs (cytostatics) are a group of antitumor drugs that disrupt and slow down the mechanisms, division, growth and development of all cells of the body, which is especially important in the presence of malignant (cancerous) tumors. Also effective for nephritis.
Good effectiveness in therapy should be given to the simultaneous use of cytostatics with glucocorticoids, especially in cases of treatment of vasculitis such as ANCA, urticarial, hemorrhagic, cryoglobulinemic, giant cell arteritis, Takayasu arteritis. The duration of taking cytostatics ranges from 3 to 12 months.
Among the cytostatics we can highlight: Cyclophosphamide, Methotrexate, Doxorubicin, Fluorouracil.
Monoclonal antibodies are antibodies produced by immune cells that have immunosuppressive and antitumor properties and have been shown to be effective against skin cancer (melanoma), breast cancer and lymphatic leukemia. Medicines from the group of monoclonal antibodies are no less effective than cytostatics and are used in the treatment of ANCA vasculitis. The appointment is advisable in case of undesirable use of cytostatic drugs. Contraindications for use are the presence of hepatitis B virus (HBV), a positive intradermal tuberculin test, neutropenia, as well as low levels of IgG (immunoglobulins class G) in the blood.
Among the monoclonal antibodies against vasculitis are: Rituximab.
Immunosuppressants are a group of drugs that suppress the action of the immune system. Prescribed in combination with glucocorticoids.
Among the monoclonal antibodies against vasculitis are: “Azathioprine”, “Mycophenolate mofetil”.
If there are contraindications to Azathioprine, Leflunomide may be prescribed.
"Mycophenolate mofetil" is prescribed as an alternative treatment for patients with refractory or recurrent systemic vasculitis, for example, with kidney damage, however, with an increase in ALT and AST in the peripheral blood by 3 times or more, as well as a decrease in platelets (100×109/l ) and leukocytes (2.5×109/l), the drug is stopped.
Normal human immunoglobulin - prescribed for severe kidney damage, infectious complications, hemorrhagic alveolitis.
Anti-infectious therapy – used for diseases of infectious etiology or concomitant infectious diseases.
If bacteria are present, antibacterial drugs are prescribed - Trimethoprim, Sulfamethoxazole.
Read also Enteritis - symptoms, causes, types and treatment of enteritis
If viruses are present, antiviral drugs are prescribed - Interferon alfa, Vidarabine, Lamivudine.
For hepatitis, drugs are prescribed by a hepatologist, depending on the type of hepatitis virus.
Detoxification therapy – used to bind toxins and biologically active substances, as well as remove them from the body.
The following detoxification drugs can be identified: Atoxil, Nutriclins, Polyphepan, Enterosgel.
Non-steroidal anti-inflammatory non-steroidal drugs (NSAIDs) - are prescribed for persistent and necrotic lesions, thrombophlebitis and nodular vasculitis.
Among NSAIDs one can highlight: Acetylsalicylic acid, Ibuprofen, Indomethacin, Phenylbutazone.
Anticoagulants are a group of drugs that prevent the formation of blood clots in blood vessels.
Anticoagulants include Warfarin and Heparin.
Antihistamine therapy is used if the patient is allergic to medications or foods.
Antihistamines include Diazolin, Claritin, Tavegil, Terfen, and Fenkarol.
Treatment of vasculitis in children also includes transfusion therapy.
Can also be used:
- Hemosorption;
- ACE inhibitors;
- Vasodilators.
2. Physiotherapeutic methods for treating vasculitis
Plasmapheresis – helps improve kidney function, reducing the risk of developing end-stage renal failure.
Diet for vasculitis
The diet for vasculitis is hypoallergenic, which is important if the disease is of an allergic nature.
Products from the highly allergenic group are excluded from the diet - eggs, whole milk, seafood (shellfish), caviar, strawberries, wild strawberries, bananas, melon, mango, persimmon, citrus fruits, tomatoes, carrots, bell peppers, nuts, coffee, cocoa, chocolate, baked goods , industrial canned food, as well as individually intolerable food products.
In case of severe nephritis, diet No. 7 is prescribed, with a further gradual transition to a hypochloride diet. For abdominal syndrome, the use of diet No. 1 is indicated.
Diagnosis of the disease
To make an accurate diagnosis, a blood test for antineutrophil cytoplasmic antibodies should be done. In 50% of cases, myeloperoxidase ANCA is detected in the patient, and proteinase-3 ANCA is detected in 40% of cases. And only in 10% of cases the result is negative.
Next, to confirm or refute the diagnosis, you need to do the following:
- general blood and urine tests;
- CT or MRI of the affected organ;
- X-ray contrast angiological examination;
- biopsy of the lungs or kidneys (depending on the suspicion of the target organ).
Differential diagnosis is extremely important, during which one distinguishes between hepatitis B and Churg-Strauss syndrome (in the first case, asthma is absent, in the second, it is present). It is also necessary to exclude other pathologies in which renal-pulmonary syndrome is noted. In particular, these are Goodpasture's syndrome, systemic vasculitis, periarteritis nodosa, hemorrhagic vasculitis, systemic lupus erythematosus, angiocentric malignant lymphoma, malignant formations, syphilis and leprosy, mycosis, tuberculosis, berylliosis, etc.
Important: With hepatitis B, antibodies to proteinase-3 are detected in the patient’s blood, while with EGPA and MPA, the patient’s blood has an increased concentration of antibodies to myeloperoxidase.
Treatment of vasculitis with folk remedies
Important!
Before using folk remedies against vasculitis, be sure to consult your doctor! Collection 1. Mix 4 tbsp. spoons of carefully crushed elderberry flowers, Japanese sophora fruits, stinging nettle leaves, yarrow grass and knotweed. 1 tbsp. Pour a glass of boiling water over a spoonful of the mixture, leave the product for an hour, and strain. The infusion should be taken during the day, 2-3 times.
Collection 2. Mix 3 tbsp. spoons of calendula flowers, elderberry flowers, mint leaves, yarrow herb, horsetail, string and poplar buds. 1 tbsp. Pour a glass of boiling water over a spoonful of the mixture, cover the container and let the product brew for 1 hour, strain. The infusion should be taken 100 ml during the day, every 3 hours.
Bergenia thickleaf. Used to cleanse the blood. To prepare the product you need 2 tbsp. Place spoons of dry bergenia leaves into a thermos and pour a glass of boiling water. The product needs to brew overnight, then filter it, add 1 tbsp. a spoonful of honey and drink it in the morning, on an empty stomach.
Differential diagnosis
During this type of diagnosis, doctors exclude other diseases and confirm the presence of Wegener's granulomatosis. Considering that this disease has a large number of diverse and common symptoms, the list of similar diseases is quite large:
- other types of vasculitis;
- malignant tumors of internal organs;
- median granuloma of the nose;
- sarcoidosis;
- lymphatoid granulomatosis;
- tuberculosis;
- syphilis;
- AIDS;
- systemic fungal diseases, etc.
Detecting a disease and making a correct diagnosis is the main task of any attending physician.
Prevention of vasculitis
Prevention of vasculitis is necessary not only to prevent the development of this disease, but also for its therapeutic purposes, which will help speed up recovery and minimize the development of complications.
Preventive measures include:
- Avoiding stress;
- Do not use medications without consulting a doctor;
- Do not leave various diseases to chance, so that they do not become chronic, especially infectious ones;
- Quitting bad habits – smoking, drinking alcoholic beverages;
- Maintaining normal body weight;
- Lead an active lifestyle, move more;
- Try not to eat unhealthy and unhealthy foods;
- Eating mainly food enriched with vitamins and microelements (minerals).
The first symptoms of granulomatosis
At the earliest stage of the disease, when it just begins to spread throughout the body, the symptoms of Wegener's granulomatosis are very similar to those of a common cold. It is for this reason that disorders of the autoimmune system are very difficult to identify in the initial stages.
- A person's body temperature rises.
- Aching pain appears in the muscles and joints.
- There is general weakness of the body.
- Lost appetite.
- Breathing becomes difficult, and signs of diseases such as rhinitis or sinusitis appear.
- Hearing disorders such as otitis media and blockage of the auditory tube are also observed.
If all of the above symptoms are present, a thorough diagnosis is necessary. This is necessary in order to more accurately determine the presence of a particular disease and begin correct and effective treatment in a timely manner.
Treatment methods
The therapeutic course regimen is drawn up individually, based on the stage of the autoimmune process, the location of the inflammatory focus, the general condition of the patient and other factors. The main goal of therapy is to achieve stable and long-term remission.
Treatment involves the use of hormonal drugs, which at an early stage help to neutralize negative symptoms. Immunosuppressants are recommended to suppress the activity of the immune system.
The classic version of the treatment regimen is as follows:
- Prednisolone + Cyclophosphamide (prescribe the maximum dose).
- As you feel better, the dose is reduced by 0.25 mg.
- Then there is a similar dosage reduction of 25 mg every 90 days, provided that the patient's condition is stable and satisfactory.
Treatment of the disease is associated with certain risks. The drugs have many side effects and suppress the immune system, which leads to a decrease in the body’s ability to resist viral and infectious processes. With prolonged use, glucocorticosteroids weaken bones.
The main recommendation of doctors is the use of glucocorticosteroids in high doses.
Against the background of local vascular damage, the dosage is the maximum possible. Drugs – Prednisolone or Methotrexate.
The course varies from 4 to 6 weeks, after which the dose is reduced.
For the generalized form, Cyclophosphamide is added to one of these medications. And the course of therapy is at least 6 months.
At this stage, it is recommended to use medications that suppress the immune system. Otherwise, the autoimmune disease will progress. The following medications are prescribed in maintenance dosages:
- Cyclophosphamide.
- Prednisolone.
- Methotrexate.
- Azathioprine.
If the effect of use is insufficient, then cyclosporines can be prescribed, but their pharmacological effect on Wegener's granulomatosis has not been fully studied. With proper treatment, remission can be achieved in 6-12 months.
In case of exacerbation, medications are used that are used at the stage of induction of remission. Most often, relapse leads to surgical intervention. If the organs of visual perception are damaged, the granuloma is resected to prevent the spread of the process and blindness of the patient. Surgery is indicated if there is a high risk of patient disability.
Modern classifications of systemic vasculitis
Most modern classifications of systemic vasculitis are limited to listing their nosological forms, observing, however, the principle of their division into primary and secondary angiitis and according to the caliber of the affected vessels [E.N. Semenkova, 1988; 1993;
N.P. Shilkina, 1988; JTLie, 1988; 1991; 1994;1996; J. C. Jannette et al., 1994; PABacon, 1996] (Table 1.6; 1.7). In addition, a number of classifications distinguish vasculopathies separately. Table. 1.6. Classification of systemic vasculitis (J. C. Jannette et al., 1994)
| Vasculitis of large vessels | Vasculitis of medium-sized vessels | Vasculitis of small vessels |
| 1. Giant cell (temporal) arteritis 2. Takayasu arteritis | 1. Polyarteritis nodosa (classic) 2. Kawasaki disease | 1. Wegener's granulomatosis 2. Churg-Strauss syndrome 3. Microscopic polyangiitis (polyarteritis) 4. Heioch-Schönlein purpura 5. Essential cryoglobulinemic vasculitis 6. Cutaneous leukocytoclastic vasculitis |
Table 1.7. Classification of systemic vasculitis (JTLie, 1994)
| Primary vasculitis | Secondary vasculitis | Vasculopathies (pseudovasculitis) |
| I. Vasculitis with predominant damage to vessels of large, medium and small caliber 1. Takayasu arteritis 2. Giant cell (temporal) arteritis 3. Isolated angiitis of the central nervous system II. Vasculitis with predominant damage to medium and small vessels 1. Polyarteritis nodosa 2. Churg-Strauss syndrome 3. Wegener's granulomatosis III. Vasculitis with predominant damage to small-caliber vessels 1. Microscopic polyangiitis 2. Schönlein-Heioch purpura 3. Cutaneous leukocytoclastic vasculitis IV. Mixed conditions 1. Thromboangiitis obliterans (Buerger's disease) 2. Cogan's syndrome 3. Kawasaki disease | 1. Infectious angiitis 2. Vasculitis in rheumatic diseases 3. Drug-induced vasculitis 4. Vasculitis in mixed cryoglobulinemia 5. Vasculitis associated with tumors 6. Hypocomplementemic urticarial vasculitis 7. Vasculitis in organ transplantation | 1. Vascular lesions in myxoma 2. Vascular lesions in endocarditis 3. Sneddon syndrome |
Visual diagnostic methods
Chest X-ray
Nonspecific abnormalities that may be seen on chest x-ray include infiltrates, nodules, macular consolidation, pleural effusion, and cardiomegaly (enlarged heart). These findings can occur in many situations, but if unexplained, may raise suspicion of vasculitis.
Angiography
Angiography may show vascular occlusion and aneurysm. The diagnosis of polyarteritis nodosa can be confirmed by the detection of aneurysms in the mesenteric and renal arteries. Although conventional angiography is still accepted as the recommended diagnostic modality, computed tomography and magnetic resonance angiography (CT and MRI) may be more effective as they can provide valuable information regarding intraluminal pathology and vessel wall thickening. These methods have been used to diagnose and monitor Takayasu's arteritis and Kawasaki disease.
Echocardiography (ultrasound of the heart)
Transthoracic echocardiography reveals coronary artery pathology in Kawasaki disease. Approximately 40 percent of children with Kawasaki disease have coronary artery disease (ectasia or aneurysm), which is detected by ultrasound of the heart. Echocardiography is used to evaluate coronary artery blood flow and the degree of coronary stenosis in patients with such arteritis.
Ultrasound echography
Ultrasonography may be useful in diagnosing and monitoring large vessel vasculitides. Patients with giant cell arteritis may have signs of stenosis, occlusion, or a halo (a dark area around the artery due to swelling of the vessel wall) of the superficial temporal artery.
CT scan
Computed tomography has diagnostic value in patients with sinonasal Wegener's granulomatosis. Findings include thickening of the nasal mucosa and focal bone destruction, mainly in the midline. Nodules or masses may be visible on a CT scan of the chest in about 90 percent of patients with Wegener's granulomatosis.
Drug treatment
Treatment of Wegener's granulomatosis is primarily aimed at achieving remission and maintaining this condition. The most commonly used drugs are:
- Corticosteroid hormones (“Prednisolone”, “Methylprednisolone”) - these medications are used at a very early stage, with their help you can suppress the aggression of the immune system towards your own cells. When taken long-term, such medications have a negative effect on bones.
- Immunosuppressants (“Cyclophosphamide”, “Azathioprine”, “Methotrexate”) - such drugs are the main ones in the treatment of Wegener’s disease. Their only drawback is that they suppress the immune system and reduce the body's resistance.
- Experimental drugs are prescribed when other treatments have failed. The most commonly used drug is Rituximab, which can be used to reduce the number of lymphocytes that play an important role during the inflammatory process.
It is worth remembering that a specific drug, its dosage and method of administration are prescribed exclusively by a specialist, taking into account all the features of the course of the disease.
Instrumental treatment methods
If Wegener's granulomatosis has reached an advanced stage and serious complications arise, the patient is prescribed various types of surgical interventions, for example:
- in case of stenosis of the respiratory tract, a tracheostomy is installed, that is, a tube that is inserted through an incision into the trachea and fixed to the skin;
- for inflammation of the nasal passages and sinuses, daily rinsing with an isotonic solution is prescribed;
- If kidney function is impaired, a donor organ transplant may be required.