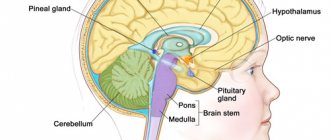
Symptoms of bacillary angiomatosis
Depending on the location, there are several types of this disease:
1. Skin.
The most common variety. Bacillary angiomatosis of the skin is characterized by the formation of painful small red-violet papules. They gradually grow and turn into nodules or rise above the surface of the skin like a Peruvian wart. At first the surface of the papules is smooth, later it becomes hemorrhagic with small areas of necrosis. Since papules affect the blood vessels of the skin, they bleed when damaged. Sometimes cellulite plaques occur.
With a subcutaneous disease, nodular plexuses with a diameter of several centimeters are formed. They are located in the layers of the epidermis on any part of the body, including the back and head. In the absence of the necessary treatment, subcutaneous nodes grow on the surface of the skin, becoming loose, and can become infected.
2. Bone.
The bone type of the disease usually precedes the skin type. The lesion consists of destruction of bone tissue and most often affects long tubular bones (radius, femur, fibula). Bacillary angiomatosis of the skull, vertebrae and ribs occurs. Bone lesions occur in one or more places. The main symptom of this type of bacillary angiomatosis is pain in the limbs.
3. Gastrointestinal.
With the gastrointestinal type, angiomatous papules form on the mucous membrane of the stomach, small and large intestines. As a result, the mucous membrane becomes hilly.
4. Extracavitary.
The extracavitary type is characterized by the presence of a large intra-abdominal formation growing into the intestine, which leads to bleeding in the gastrointestinal tract. As a result of the enlargement of the abdominal, peripancreatic and portohepatic nodes, compression of the main bile duct occurs and the development of mechanical jaundice.
5. Respiratory.
This type is characterized by the formation of polypoid formations in the bronchi and trachea. In some cases, laryngeal obstruction occurs.
Common symptoms for any type of bacillary angiomatosis are fever, malaise, headache, vomiting, and decreased appetite. When the infection generalizes, encephalitis, aseptic meningitis, chronic dysfunction of the central nervous system and mental disorders develop.
Symptoms of angiomatosis:
Clinically, it is a local process without a tendency to progress. Systemic angiomatosis (multiple, diffuse) is characterized by the appearance of multiple angiomas, vascular spots, telangiectasia in various organs and tissues. The process often takes on a progressive nature, the affected area increases, and new lesions appear. For example, the process can involve the entire limb and disrupt its function. Systemic angiomatoses include Randu-Osler disease, a group of congenital diseases (eg, Hippel-Lindau disease, etc.). With this form of angiomatosis in the skin, soft tissues, bones, blood vessels of a large caliber grow with the formation of cavernous cavities, trophic ulcers develop, bleeding occurs, etc. The course of the disease is chronic. The prognosis depends on the location and size of the lesion.
STURGE-WEBER-KRABBE SYNDROME
STURGE-WEBER-KRABBE SYNDROME
(syn. encephalotrigeminal angiomatosis) is a congenital symptom complex of abnormalities of the blood vessels of the brain, skin and eyes, characterized by unilateral angioma of the facial skin in combination with leptomeningeal angiomatosis on the same side. The syndrome is based on a defect in the development of blood vessels in the skin, brain, and often the eyes, possibly due to a violation of vascular innervation. An autosomal dominant type of inheritance is assumed; cases of trisomy on the 22nd chromosome have been described. Skin lesions such as flaming nevus are observed from birth. The nevus is usually located on one side of the facial skin in the area of the first and second branches of the trigeminal nerve. The size of an angioma can be several centimeters, but more often it tends to spread to the scalp, neck, and sometimes even to the torso. Often, angioma affects the forehead, eyelids, nose, lips and mucous membrane of the nose and mouth on the affected side. In 40% of cases, facial damage can be bilateral, but even in this case, one half is affected to a much greater extent. The color of the angioma is bright or dark red, the surface is smooth, in rare cases the angioma is cavernous and hypertrophy of soft tissues and bones develops in its area. Histologically, ectasia of thin-walled vessels of the superficial plexus of the dermis, the lumens of which are filled with red blood cells, is usually detected. Neurological symptoms appear more often in early childhood; in very rare cases, leptomeningeal angiomatosis remains asymptomatic throughout life. In 75-90% of cases, it manifests itself as epilepsy (usually already in the 2-7th month of life), hemiparesis, and mental retardation. X-rays often reveal cortical calcification in the form of convoluted, ribbon-like stripes that follow the contours of the surface of the brain. Using computed tomography after administration of contrast agents and magnetic resonance, the intracranial localization of angiomatosis is clarified. Eye damage is observed in 30-60% of cases in the form of angiomatosis of the conjunctiva, choroid, glaucoma, which can lead to loss of vision. The prognosis is unfavorable, the course of the disease is progressive, worsens with skull injuries, infections, and death is possible at a young age. The diagnosis is made on the basis of clinical radiological, computed tomographic, and angiographic data. Differential diagnosis is carried out with other types of angiomas.
Treatment is symptomatic. Individual foci of skin angiomatosis can be removed using cryosurgery, surgical excision followed by plastic surgery, or laser therapy.
General information
Sturge-Weber syndrome is a rare congenital angiomatous lesion of the cerebral membranes, skin and eyes.
The prevalence is at the level of 1 case per 100 thousand population. For the first time, a patient with such a syndrome was described in 1879 by Sturge, then in 1922 by Weber, who indicated the radiological signs detected in this syndrome. In 1934, Krabbe suggested that patients, along with skin angiomas, have angiomatosis of the cerebral membranes. In honor of these researchers, the disease was named Sturge-Weber-Krabbe syndrome. Due to the fact that facial angiomas are localized in the area of skin innervation by the 1st and 2nd branches of the trigeminal nerve, in neurology the disease is also known as encephalotrigeminal angiomatosis.
Sturge-Weber syndrome
Types of angiomatosis
Vascular changes can occur in the liver, face, respiratory, and digestive organs. In addition, the disease can affect the skin, bones, nervous system
Please note that the local form of the disease is not dangerous to human life, it is benign
Scientists are still trying to unravel the reasons for the development of the syndrome. Most of all, scientists are inclined to believe that the disease is triggered by toxic influences when tissues and organs are formed, as well as gene mutations.
Often the pathology is caused by the fact that the expectant mother:
- He is fond of alcohol and drugs.
- Smokes a lot.
- Uses illegal drugs.
- Has extragenital pathology of the thyroid gland and diabetes mellitus.
Signs of the disease depend on where the disease is located. If the central nervous system is affected, it ends in blindness and dysfunction of internal organs.
The prognosis in most cases depends on the form of the disease. Most often it is favorable, but it happens that the disease progresses significantly and ends in severe disability.
The following types of angiomatosis can be distinguished:
- Diffuse – accompanied by systemic damage.
- Isolated – the retina is affected.
- Bacillary occurs in case of bacterial infection.
Sturge-Weber syndrome affects the eye vessels, facial skin, and lining of the brain. The pathology is considered congenital and quite rare. It is dangerous that the syndrome affects the eyes and brain - angiomas appear here, leading to disability.
The classic version is characterized by damage to the nervous system, eyes, and the appearance of bright red spots on the face. The disease can occur in the following ways:
- Skin angioma, leading to glaucoma, damage to the membranes of the brain.
- Damage to the nervous system.
Typically, one part of the face is affected. The spot is located under the eye on the forehead and can go down to the lip. When you press on the skin, it turns pale. In newborns, the spot on the face is pink, after a while it becomes cherry. Gradually, the formation begins to increase and completely affects one half of the face.
In addition to vascular disorders, you can notice the following manifestations:
Types of pathology
The typical combination of skin, eye and nervous system lesions is not present in all patients. In approximately 80% of those suffering from Weber's angiomatosis, the combination of symptoms is not so obvious. In addition, similar symptoms may occur with other congenital anomalies of vascular structure.
Focal
In the case of isolated skin lesions, patients can be completely socially adapted, since they have only cosmetic defects. Vision is not impaired, and there are no abnormalities in the examination of the nervous system.
On the skin and retina
Refers to relatively favorable variants of Weber syndrome, since there are no neurological symptoms. Such patients may not experience problems with mental development, and the course of the disease will depend on the rate of progression of glaucoma.
Bacillary
Occurs with immunodeficiency. The causative agent of the disease, rickettsia, is transmitted to humans through cat scratches. This type of angiomatosis is characterized by the appearance of nodular rashes throughout the skin of the body. One patient may have more than 1000 of them. These papules are not painful; when punctured, intense bleeding occurs.
Hemorrhagic
Areas of dilated vessels form on the wings of the nose; most often, the first manifestations occur in school-age children. Subsequently, the area of angiectasias increases; they can be noticeable on any part of the skin and mucous membranes. Randu-Osler-Weber syndrome is accompanied by nosebleeds that are mild at first and then can become profuse and extremely persistent.
Lasting for several weeks, hemorrhages lead to anemia and can be fatal.
Arteriovenous
Neuroretinal angiomatosis of Wyburn-Mason occurs with the formation of direct communications between the arterial and venous networks. Vascular malformations occur in the choroid of the eyes and brain. But, unlike Weber syndrome, patients do not suffer from glaucoma. The examination reveals changes in the fundus, loss of visual fields, impaired thinking, and damage to the cranial nerves.
Watch the video about the symptoms of Weber's angiomatosis and treatment:
Characteristic symptoms of the disease
The pathology belongs to an alternating group of syndromes that extend to the cranial, facial and eyeball nerves. The neurological disorder leads to partial loss of motor function on the side opposite to the lesion, to the development of strabismus, paralysis of the muscles of the tongue and face. The main indicator of the disease is a burgundy spot on one side of the face (photo) or torso; less commonly, capillary hemangioma has a bilateral localization.
Based on the nature of the syndrome, three combinations of signs are determined:
- vascular angioma of the intracranial membranes;
- angiomatosis of the face with damage to the retina;
- combination of capillary tumors (brain, face, eyes).
Each form is accompanied by corresponding anomalous disorders.
Facial lesion
The manifestation of the syndrome on the facial part is called “flaming nevus”, is present at birth or develops later. Angioma has the shape of a spot with blurred edges and changes with the proliferation of capillaries. The neoplasm is bright red or burgundy in color, covers half the forehead, one side of the nose, the upper cheekbone, the eye area, and ends above the upper lip. This is a classic symptom of the disease; “angel's kiss” can appear on both sides, covering the scalp, the back of the head and down to the torso.
Foci of localization of vascular lesions can be single, scattered or merging into a common form. When pressed, the problem area turns pale, then the color is restored. Over the course of a person’s life, the “flaming nevus” changes not only its shape, but also its color. In a newborn, this spot is light pink in color; as it grows, it darkens, acquiring a burgundy hue.
In most cases, the angioma is smooth and does not protrude above the surface of the skin. In a more complex form, the anomaly is accompanied by pigmentation, moles, limited edema, and hemangioma. Clearly expressed symptoms on the face are diagnosed in 99% of cases.
Neurological changes
Angiomatosis of brain tissue leads to calcification of the capillaries of the affected area, which, in turn, provokes the development of such pathologies:
- intracranial atrophy and necrosis;
- increase in glial cells;
- hydrocephalus;
- dementia.
Signs of brain damage begin to appear in the first year of a child’s life. The disease is characterized by a long clinical course, in which the signs of a neurological disorder progress, causing impaired motor function and mental retardation.
The proliferation of blood vessels creates pressure on neurons, creating a convulsive syndrome. A pattern is noted: the greater the mental retardation, the more pronounced the epileptic seizures. Muscle contraction is activated on the side opposite to the lesion. Over time, the area of capillary proliferation increases, and attacks progress. These factors affect the emotional state and cause mental deviations in behavior. Sturge-Weber syndrome is accompanied by the following symptoms:
- cognitive disorders;
- memory impairment;
- decreased learning ability;
- irritability;
- aggression;
- rancor, vindictiveness.
In adults, each epileptic seizure leads to a decrease in intelligence. Intracranial pressure causes constant headaches. Paresis, paralysis, muscle weakness, and loss of sensitivity are formed on the side opposite to the brain lesion.
Eye signs
Ophthalmic changes associated with the disease are characterized by abnormal changes in the vascular tract. The angioma is localized in the papillomacular part. A light gray circle-shaped neoplasm (photo) calcifies, creating pressure and a favorable environment for the development of glaucoma.
The pathology disrupts the flow of fluid inside the eye, and hydrophthalmos is formed. Sturge-Weber syndrome in ophthalmology is manifested by the following problems:
- corneal clouding;
- enlargement of the eyeball;
- decreased vision (complete blindness is possible);
- narrowing the boundaries of visual capture;
- retinal atrophy.
Glaucoma may appear immediately after birth or develop later.
Symptoms of Sturge-Weber disease
Depending on the combination of manifestations of the pathology, several clinical options are conventionally distinguished:
- Angiomas of the facial skin are combined with damage to the meninges; glaucoma is optional.
- Isolated facial lesion in the absence of angiomas within the skull, glaucoma is possible.
- Isolated damage to the nervous system, glaucoma is not typical.
Facial lesion
Usually one half of the face is affected, the spot is located in the area of action of the I-II branches of the trigeminal nerve, so the redness is expressed under the eye, in the cheekbone area, on the forehead, going down to the upper lip. Foci of angiomatosis can be scattered, single or merging, then they speak of a “flaming nevus”. Due to the vascular nature of the changes, when pressure is applied to the skin, it will turn pale like other types of angiomas.
At birth, the spot on the facial part of the skin may be pink, but over time it will acquire a brighter color, turning red or cherry - a “wine stain”. A gradual increase in the size of the formation is possible. Up to 70% of patients have damage to one half of the face, almost half suffer from facial angiomas in combination with vascular anomalies of the body and limbs.
In addition to vascular skin lesions, possible manifestations of Sturge-Weber angiomatosis include moles, skin pigmentation disorders, limited soft tissue swelling, and true vascular tumors (hemangiomas). Pathology is very rarely diagnosed without a characteristic lesion of the facial skin.
Neurological changes
Angiomatosis of the pia mater is the most important component of the syndrome, determining the severity of the course and poor prognosis. Signs of damage to the nervous system appear already during the first year of a child’s life and steadily progress, causing severe mental retardation and impaired motor and sensory function.
example of severe brain damage a. Sturge-Weber in a 2-year-old child
Clinical signs of angiomatosis of the meninges:
- Convulsive syndrome - generalized convulsions, contractions of the limbs opposite to the affected half of the brain, absence seizures (short-term attacks without convulsions, when the patient seems to freeze, “absent” for several seconds or minutes);
- Disorders of mental development and behavior;
- Hemiparesis and paralysis on the side opposite to the site of brain damage, weakness of the muscles of the limbs;
- Decreased skin sensitivity on the side opposite to the lesion of the meninges.
Neurological symptoms are associated with the impact of vascular growths in the pia mater on the cerebral cortex, irritation of the neurons of which causes seizures. Increased intracranial pressure provokes frequent migraine-type headaches.
Compression of areas of the brain contributes to necrosis, atrophy, proliferation of glial elements (similar to sclerosis), and deposition of calcium salts, which significantly disrupts brain function up to dementia. Congenital angiomatosis of the meninges can cause hydrocephalus.
To a large extent, the emotional sphere suffers with Sturge-Weber syndrome. Patients acquire characteristic psychopathological personality traits: they are tearful, often and unreasonably irritated, prone to aggression, vindictive, self-centered, which significantly affects the process of education and communication.
As intelligence declines, memory and attention suffer; children’s education can become extremely difficult or impossible due to imbecility or idiocy. In adults, there is a gradual decline in intellectual abilities, progressing from seizure to seizure
Changes in behavior can also be observed in the period between seizures.
A dangerous manifestation of angiomatosis of the soft shell of the brain can be status epilepticus, when convulsive seizures follow one after another, preventing the body from at least partially recovering. This condition requires emergency care and can lead to brain swelling and death of the patient.
Eye symptoms
Eye damage consists of the formation of angiomas of the vascular tract of the eye, which leads to an increase in intraocular pressure and glaucoma.
Vascular conglomerates disrupt the normal flow of intraocular fluid, and hydrophthalmos occurs. Glaucoma can be expressed immediately after birth or develop later. Damage to eye tissue is accompanied by a progressive decrease in visual acuity and loss of its fields. With glaucoma, there is a high probability of retinal atrophy and detachment, which can lead to complete loss of vision.
ENCEPHALOTRIGEMINAL ANGIOMATOSIS
ENCEPHALOTRIGEMINAL ANGIOMATOSIS
(synonym:
Sturge-Weber disease, Sturge-Weber-Krabbe syndrome, Sturge-Weber-Krabbe-Dimitri angiomatosis, neuroectodermal congenital dysplasia
) is a disease from the group of phakomatoses, which is based on ectomesodermal dysplasia, manifested by angiomatosis of the skin, brain, eyeball, and sometimes also angiomatosis and malformations of internal organs.
Encephalotrigeminal angiomatosis was first described in detail in 1879 by the English ophthalmologist WA S turge, who observed a patient with angioma of the facial skin, glaucoma, seizures and hemiparesis. In 1922, Weber (FP Weber) discovered intracranial petrification on a radiograph of a patient with similar clinical manifestations. Similar radiological findings were reported by V. Dimitri in 1923, Krabbe (K. H. Krabbe, 1934) described in detail the morphological changes in this disease. In 1937, encephalotrigeminal angiomatosis was included by J. van der Hoeve in the group of phakomatoses (see Phakomatoses).
In the development of encephalotrigeminal angiomatosis, the leading role appears to be played by genetic factors, which is confirmed by the presence of familial forms of the disease, as well as the high concordance (similarity) of the course of the disease in monozygotic twins. The type of inheritance is autosomal dominant with incomplete penetrance, autosomal recessive is possible.
The disease is based on hereditary disorders that lead to ectomesodermal dysplasia with the development of angiomatosis (see Angiomatosis) of the skin, brain, retina and choroid, and sometimes internal organs. Hemangiomas (see) of the brain are located mainly on the surface of the cerebral hemispheres in the parietal and occipital lobes, often on the same side as angiomas of the facial skin. Hemangiomas can spread to the deep parts of the brain, occupying almost the entire hemisphere. With diffuse angiomatosis of the skin, pronounced angiomatosis of the brain is not always observed. Angiomatosis of the eyeballs is characterized by sclerosis and atrophy of the iris, ciliary body, and retinal detachment. There is also a thickening of the choroid, containing a large number of dilated blood vessels. Sometimes angiomas and malformations of internal organs are detected.
The earliest clinical manifestations of the disease are skin hemangiomas, which can be observed from birth or appear in the first months of a child’s life. Skin hemangiomas have different shapes and sizes, usually bright red or dark cherry color. They are more often located on the face on one side in the zone of innervation of the trigeminal nerve (mainly the first and second branches), less often they are bilateral; can also be localized on the trunk and limbs. Hypertrophy and swelling of soft tissues and mucous membranes are often observed. In addition, coffee-colored spots and areas of hypopigmentation are found on the skin of the trunk and limbs (see Vitiligo). Early manifestations of encephalotrigeminal angiomatosis also include focal epileptic seizures (see Epilepsy). In children of the first years of life, tonic convulsions predominate. In school-age children and adults, focal motor, sensory, vegetovisceral and combined attacks are observed. There is no correlation between the prevalence of cutaneous angiomatosis and the severity of epileptic seizures (the latter are caused by the presence of intracranial angiomas and hemocerebrospinal fluid-dynamic cerebral disorders). The child's mental development is lagging behind. A decrease in intelligence can reach the level of imbecility, less often idiocy. There is a correlation between the degree of mental degradation and the severity of epileptic seizures. In the interictal period, motor disinhibition and aggressiveness of patients are noted. Hemiparesis and hemiplegia often occur before or shortly after the onset of seizures and are characterized by persistence. Less commonly observed are hyperkinesis (see), muscle dystonia, locomotor and static ataxia (see), damage to the V, VI, VII pairs of cranial nerves, hypothalamic disorders (vegetative-trophic, endocrine-metabolic, accompanied by premature sexual development). The severity of cerebral and focal neurological disorders is very variable.
An ophthalmological examination reveals angiomatosis of the conjunctiva, iris, ciliary body and choroid. Less commonly, the retina and optic nerve head are involved in the process. Angiomatosis of the choroid progresses and leads to glaucoma and blindness.
The course of the disease is progressive, central paralysis gradually increases, epileptic seizures become more frequent, and severe mental disorders develop. Remissions may occur, during which epileptic seizures stop. In addition to the described typical wedge, forms of the disease there are abortive forms with the absence of any one or group of symptoms (hemiparesis, glaucoma, epileptic seizures, etc.).
The diagnosis of encephalotrigeminal angiomatosis is based on identifying a complex of characteristic wedges, manifestations and examination data. X-rays of the skull reveal petrification and signs of intracranial hypertension. An electroencephalographic study reveals focal or diffuse changes of an epileptic nature. Cerebrospinal fluid is usually unchanged. Differential diagnosis is carried out with congenital malformations of the central nervous system (see Brain), other diseases from the group of phakomatoses (see).
Treatment of encephalotrigeminal angiomatosis is symptomatic. Antiepileptic drugs (see), dehydration therapy (see) are used. Surgical treatment of angiomas of the skin and brain is carried out for limited, isolated lesions. Radiation therapy is sometimes indicated for cerebral hemangioma. With glaucoma, in some cases, enucleation of the eye is performed (see).
The prognosis is unfavorable. As encephalotrigeminal angiomatosis progresses, patients die (death occurs during status epilepticus from cerebral edema). Infectious diseases and injuries can aggravate the course of encephalotrigeminal angiomatosis and often lead to death due to the development of subarachnoid-parenchymal hemorrhages.
Prevention involves medical genetic counseling (see Medical genetic consultation) with a recommendation in some cases to limit childbearing in families with registered cases of encephalotrigeminal angiomatosis.
Bibliography:
Arkhipov B. A., Skvortsov I. A. and Kamennykh L. N. Encephalotrigeminal Sturge-Weber angiomatosis (Clinical electroneuromyographic study), Zhurn. neuropath. and psychiat., t. 82, no. 5, p. 38, 1982; Badalyan JI. O. et al. Hereditary angiomatoses with predominant damage to the nervous system. Message 1. Sturge-Weber disease, Pediatrics, No. 9, p. 77, 1969; Voloshin V. X. and Chekanov Yu. N. Operation iridocycloretraction for Sturge-Weber-Krabbe syndrome, Vestn. ophthalm., No. 5, p. 5, 1974; Kalinina JI. V. Neurological syndromes with phakomatoses in children, Zhurn. neuropath. and psychiat., vol. 76, no. 10, p. 1487, 1976; Kondrashin N. I. Clinic and treatment of hemangiomas in children, M., 1963; Problems of clinical neuropathology, ed. JI. M. Shenderovich, p. 58, Vladivostok, 1973; In aik o ff G. e. a* Maladie de Sturge - Weber et glaucome, Bull. Soc. Opbtal. Fr., t. 80, p. 395, 1980; Czarnecki DB The Sturge — Weber syndrome, Arch. Derm., v. 117, p. 305, 1981; Feng Y. K. a. Yang Y. C* Sturge — Weber syndrome, Chin. med. J., y. 93, p. 697, 1980.
L. V. Kalinina.
Treatment and prognosis
Currently, there is no effective therapy. It is aimed at relieving the main symptoms (anticonvulsant treatment).
Episyndrome often turns out to be resistant to antiepileptic treatment, which forces a change in monotherapy to a combination of two drugs.
One of the treatment methods is X-ray irradiation of the skull over the damaged area. According to indications, surgical treatment of epilepsy is possible.
Glaucoma therapy involves the use of eye drops that reduce the secretion of intraocular fluid. But conservative treatment is often ineffective, then surgical treatment of glaucoma is indicated.
The life prognosis, especially with severe symptoms, is unfavorable. Episyndrome that cannot be stopped leads to mental retardation. Sometimes there is loss of vision, intracranial vascular disorders leading to stroke.
Diagnosis of the disease
How to determine the disease? The diagnosis of “bacillary angiomatosis” is made based on the results of the initial examination and histological examination of the biopsy specimen. How does this happen?
The initial examination allows you to identify the presence, nature and location of papules, as well as assess the severity of the disease. Histological examination of blood in bacillary angiomatosis allows one to observe the proliferation of capillaries, in which epithelial endothelial cells with mitoses and enlarged nuclei are clearly visible.
Important point. Before making a diagnosis, it is necessary to carry out differential diagnosis with a number of diseases that are similar to bacillary angiomatosis (pyogenic granuloma, Kaposi's sarcoma, angiocreatoma and others).
Diagnostics
| Diagnostics | |
| CSF examination | Increased protein content |
| X-ray of the skull | Tram rail symptom |
| Angiography | Incompetent superficial cortical veins Empty dural sinuses Pathological tortuosity of veins ( preferably MRI [1]) |
| CT scan | Calcifications, tram rail symptom Cortical atrophy Abnormal draining veins Enlarged choroid plexus Blood-brain barrier breakdown (during seizures) Contrast enhancement |
| MRI[1] | Gadolinium enhancement of LA Enlarged choroid plexus Sinovenous occlusion Cortical atrophy Accelerated myelination |
| SPECT | Hyperperfusion, early Hypoperfusion, late |
| PAT | Hypometabolism |
| EEG | Reduced background activity Polymorphic delta activity Epileptiform features |
| Biopsy | |
Therapy
And finally. Treatment of bacillary angiomatosis consists of antibiotic therapy (erythromycin, doxycycline, tetracycline, minocycline, ciprofloxacin and norfloxacin). For single lesions, local therapeutic procedures are prescribed: excision, cryotherapy or electrosurgery. If necessary treatment measures are delayed, the disease may be fatal.
The duration of treatment ranges from two weeks to several months. If relapses of bacillary angiomatosis occur (with HIV infection), therapy is lifelong. To prevent re-development of the disease, it is recommended to avoid contact with carriers of the disease, especially cats and dogs.
All materials are published under the authorship or editorship of medical professionals (about the authors), but are not a prescription for treatment. Contact the specialists!
When using materials, a link or indication of the source name is required.
Angiomatosis is understood as a condition manifested by excessive proliferation of blood vessels that acquire the characteristics of tumor growth. This syndrome can be observed in a variety of diseases, but all of them are usually severe.
Angiomatosis accompanies developmental defects, appears in people with impaired immunity, and can be combined with other congenital anomalies. Foci of proliferation of blood vessels are found in the skin, internal organs, meninges, vascular system of the eyes, etc.
Depending on the prevalence, local angiomatosis is distinguished when the lesion is limited. Typically, such a change appears in one area of the body - on the face, in the liver, in the mucous membranes of the digestive or respiratory organs. In other cases, angiomatosis manifests itself as a systemic lesion that simultaneously affects the skin, internal organs, bones, and nervous system.
Local angiomatosis usually does not pose a threat to life and proceeds benignly, while the systemic process is fraught with dysfunction of vital organs, severe neurological disorders, and bleeding.
It has not yet been possible to find out the exact cause of the tumor-like growth of blood vessels, but the role of a hereditary factor and a violation of organ formation in the process of embryonic development is assumed. Certain importance is given to immunodeficiency syndromes.
The symptoms of angiomatosis depend, first of all, on the location of the lesion. The most characteristic signs are the presence of a tumor-like conglomerate of vessels of different sizes, which can cause bleeding. If the central nervous system is damaged, severe neurological symptoms develop; if the eyes are involved, blindness is possible; with angiomatosis of the internal organs, their function is impaired.
local angioma on the face (left) and angiomatosis, which may be a sign of systemic disease (right)
Patients with angiomatosis are treated by a variety of specialists. If changes affect the skin, then the patient is referred to a dermatologist or surgeon; if the eyes are affected, the help of an ophthalmologist is needed; involvement of the brain and its membranes requires the participation of a neurologist.
Treatment of angiomatosis includes both surgical intervention and a symptomatic approach; tactics are determined by the localization and behavior of vascular lesions, so the principles of therapy vary greatly among different patients. The prognosis depends on the form of the pathology: in some cases it is quite favorable, in others the disease progresses, leading to severe disability, without giving a chance of cure.
Among the types of angiomatosis are:
- Diffuse angiomatosis, accompanied by systemic damage;
- Isolated retinal angiomatosis;
- Kaposi's angiomatosis is a manifestation of sarcoma characteristic of immunodeficiency syndrome due to HIV infection;
- Sturge-Weber angiomatosis;
- Bacillary angiomatosis, which occurs due to a bacterial infection.
It is clear that such a division is largely arbitrary, because angiomatosis can be not only an independent pathology, but also a manifestation of other diseases, as happens with AIDS, for example. One of the rare, but very serious pathologies is Sturge-Weber angiomatosis, which will be discussed further.
Classification
Today in medicine the following classification of such a pathological process is:
- Retinal angiomatosis. It is very rare and is characterized by angiomatous and cystic formation in the retina, nervous and endocrine system and other internal organs. Due to the destruction of eye tissue, vision deteriorates. Glaucoma develops, which can lead to complete loss of vision.
- Cobb's or angiomatosis affecting the skin and spinal cord.
- Bacillary angiomatosis. Plaques similar to Kaposi's sarcoma begin to appear on the skin. In addition to the skin, the disease affects internal organs, spleen, liver, and bones. It should be noted that bacillary angiomatosis has another name - hepatic.
- Bacterial angiomatosis. It manifests itself in patients with HIV infection, slightly less often in cancer cases, and affects the mucous membrane of the mouth and skin. In this case, symptoms such as increased temperature, increased sweating, abdominal pain, and anemia appear. It can be diagnosed based on the results of a histological examination. Systemic treatment with certain medications is used.
- Steiner–Ferner syndrome is a consequence of tumors developing in the body.
- Sturge-Weber angiomatosis. It also has another name - encephalotrigeminal angiomatosis. Angiomas affect the brain and facial skin. It also has an effect on the development of glaucoma.
- Diffuse (venous angiomatosis) is a vascular neoplasm that includes several angiomas. Venous-cavernous developing angiomatosis is a multi-chamber cavity of vessels filled with blood. Their temperature regime is slightly higher than the surrounding tissue. A distinctive feature of venous angiomatosis is the enlargement of the affected organ and the formation of venous cavities.
Leiomyoma is especially dangerous when its vessels begin to enlarge. This condition can lead to degeneration into a cancerous tumor. In one case or another, you should not self-medicate; you should consult a doctor.
Diagnosis
Diagnosis is possible based on a characteristic combination of signs:
- the presence of facial angiomatosis of the skin;
- epileptic seizures and other neurological manifestations;
- ophthalmological picture (especially glaucoma).
An X-ray examination of the skull reveals areas of cortical calcification with double boundaries, as if encircling the convolutions in the area of brain damage. CT scan shows large areas of calcification. MRI allows you to identify atrophied and degenerative zones in the cerebral substance, cortical thinning and differentiate other clinically similar pathologies.
EEG allows you to determine the nature of bioelectrical cerebral activity and identify epiactivity.
Ophthalmic examination includes assessment of visual acuity and intraocular pressure, perimetry, ophthalmoscopy and gonioscopy, ultrasound biometry of the eye, AV scanning
Diagnostic methods
The disease is uncommon; its individual manifestations are similar to angiomatosis in other congenital malformations. Therefore, when making a diagnosis, skin and neurological symptoms are taken into account, and when determining ophthalmological disorders, they are guided by the presence of glaucoma.
Instrumental examination includes:
- radiography - along the convolutions of the brain there are lines of calcium deposition, creating a double contour (like tram rails);
- reveals in more detail affected areas and calcifications in brain tissue;
- MRI shows areas of atrophy and thinning of the medulla under the area of angiomatosis;
- EEG – the study reveals signs of epileptic activity.
A consultation with an ophthalmologist is indicated with measurement of visual acuity and eye pressure, examination of the fundus and eye biometry using ultrasound.
Treatment
Depending on the type and location of clinical pathologies, appropriate treatment for angiomatosis is prescribed:
- effective surgical intervention - removal of the resulting tumors;
- X-ray therapy;
- cryotherapy – treatment of the body with low temperatures;
- medications against seizures and psychotropic drugs are prescribed;
- agents that restore blood vessels and stop bleeding;
- tamponation of bleeding vessels; in addition, hemophobin and a hemostatic sponge are prescribed;
- If posthemorrhagic anemia occurs, a blood transfusion procedure and the required number of red blood cells are performed.
With angiomatosis, you can often find a patient suffering from epilepsy. In this case, drug treatment is prescribed. During periods of attacks, seduxen is prescribed; for long-term use, depakine, carbamazepine, finlepsin, and valproic acid are used.
”alt=””>
It should be remembered that prescribed anti-seizure drugs do not eliminate the source of pulsations in the brain. This epilepsy often turns out to be safe in relation to conventional treatment; therefore, patients are prescribed several drugs, combining them.
There is currently no effective treatment for Sturge-Weber syndrome. Therapy is aimed at relieving the main manifestations. Anticonvulsant therapy is carried out with valproate, carbamazepine, levetiracetam, topiramate.
Episyndrome often turns out to be resistant to antiepileptic treatment, which requires a transition from monotherapy to a combination of two drugs. One of the treatment methods is X-ray irradiation of the skull over the area affected by angiomatosis. According to indications, neurosurgeons may consider the issue of surgical treatment of epilepsy.
Treatment of glaucoma consists of instillation of eye drops that reduce the secretion of intraocular fluid: brimonidine, timolol, dorzolamide, brinzolamide, etc. However, such conservative therapy is often ineffective. In such cases, ophthalmic surgeons perform surgical treatment of glaucoma: trabeculotomy or trabeculectomy.
Unfortunately, with severe clinical manifestations, Sturge-Weber syndrome has an unfavorable prognosis. Intractable epileptic syndrome leads to severe mental retardation. Possible loss of vision, intracranial vascular disorders, leading to the likelihood of developing a stroke.