Болезнь Рандю-Ослера относится к наследственным вазопатиям, точнее, ее называют диссеминированным гемангиоматозом. Особенности истончения стенок капилляров к шестилетнему возрасту ребенка вызывают изменение структуры и превращают их в мелкие кровоточащие аневризмы, что нарушает местную циркуляцию крови в тканях.
Первые данные о клинике описаны французским врачом Луи Мари Рандю, в дальнейшем более полная информация представлена сэром Уильямом Ослером и Фредериком Парксом Вебером в конце XIX и начале XX века. Поэтому полное название патологии — болезнь Рандю-Ослера-Вебера.
Выявляется у 1 человека из 5000 населения. Одинаково часто болеют как мальчики, так и девочки. Максимально проявляется в среднем возрасте (в 40–50 лет). В лечении приходится сталкиваться с повторными трудно останавливаемыми кровотечениями.
В МКБ-10 патология включена в подкласс «Болезней капилляров» с кодом I78.0.
Что известно о причинах заболевания?
До настоящего времени точная причина болезни не выяснена. Установлена связь с генетическими мутациями и передачей по наследству измененных генов по доминантному типу, при наличии у одного из родителей. Однако имеются случаи, независимые от наследования, их называют спорадическими.
Генетические исследования позволили выделить 3 типа нарушений:
- I тип — ген, отвечающий за синтез коллагена;
- II тип — дефектура рецепторов фактора роста опухоли;
- III тип — имеет сложную маркировку (600604, 12p11–p12; Â).
В результате главными источниками кровотечения являются:
- очаги истончения стенок капилляров венозного и артериального вида;
- неконтролируемое расширение диаметра микрососудов;
- артериовенозные аневризмы.
Установлено негативное влияние перенесенного будущей матерью инфекционных заболеваний в первом триместре беременности, приема лекарственных препаратов.
Провоцирующими факторами кровотечения могут быть:
- недостаток витаминизации пищи, вегетарианская диета;
- плотно прилегающая одежда, травмирующая поверхностные сосуды.
Современные представления о болезни
Современные данные позволили установить основу нарушения анатомической целостности сосудов (дисплазии). Неполноценность мезенхимальной оболочки приводит к частичному отсутствию эластичных и мышечных волокон и замене их на рыхлую соединительную ткань.
На участках венул и артериол формируются выпячивания типа аневризматических расширений (телеангиэктазов)
Легкая травмированность вызывает кровотечения. Поражение капилляров при болезни Рандю-Ослера имеет не только поверхностную кожную локализацию, но распространяется на слизистые оболочки внутренних органов, чаще всего бронхи, носоглотку, ротовую полость, мочевой пузырь.
Эффективность лечения
Терапия назначается как с учетом анамнестических данных, так и на основании результатов обследования. Тактику борьбы с патологией определяет врач. Основу лечения болезни Рандю-Ослера представляет использование медикаментозных средств, направленное на остановку уже имеющихся кровотечений и профилактику возникновения новых. С этой целью назначаются такие препараты, как «Викасол», «Этамзилат» и «Аминокапроновая кислота». Их используют как в инъекционной форме, так и для местных обработок пораженных покровов. Эти вещества способствуют нормализации реологических свойств крови.
Если у пациента выявляются дефекты крупных сосудов, например, аневризма аорты, оправдано хирургическое вмешательство. Для лечения незначительных поражений используются лазерные техники и криодеструкция.
В редких случаях пациентам требуется госпитализация. Она оправдана при значительных кровотечениях. После восстановления целостности сосудов больные проходят лечение в отделении интенсивной терапии. Проводится корректировка потери жидкости за счет инфузий. В ряде случаев требуется гемотрансфузия.
Клинические проявления
Несмотря на наследственную передачу, болезнь Рандю-Ослера начинает проявляться после 10 лет, а чаще в период полового созревания и к 40 годам.
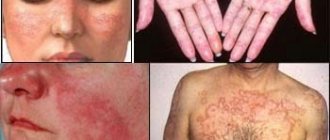
Первые признаки выражены сосудистыми «звездочками» с темно-красными папулами в центре. Они располагаются:
- на коже носа,
- на губах,
- во рту (десна, язык),
- на ушах,
- в зоне волосистой части головы.
При травмировании ангиоэктазы кровоточат. Возможен единственный признак — носовые кровотечения.
С возрастом количество высыпаний увеличивается, они распространяются на слизистые разных органов. Более всего поражаются желудок и кишечник. Поэтому к симптоматике присоединяются кровотечения из желудочно-кишечного тракта.
При разрыве аналогичных образований в оболочке головного мозга проявляется клиника инсульта. У девушек и женщин возможны маточные кровотечения, обильные менструации.
Повторные кровотечения сопровождаются развитием железодефицитной анемии. У пациентов появляются:
- слабость,
- головные боли,
- головокружение.
Промежуточную форму телеангиэктазий отличают сосудистые пачки вокруг пятна в центре
Ослер выделил 3 разновидности телеангиэктазий:
- ранние — имеют форму маленьких пятнышек с неправильными очертаниями;
- промежуточные — характерны сосудистые отростки (анастомозы между артериолами и венулами);
- узловатые — образуются яркие красно-синюшные узелки в центре высыпаний, выступающие над кожей на 1–3 мм, размер их диаметра доходит до 5–7 мм.
У пациентов после 25 лет имеются ангиэктазии двух или всех типов. Чтобы отличить их от других сосудистых образований, нужно слегка надавить на поверхность, пятно побледнеет и снова наполнится кровью после остановки контакта.
Телеангиэктазии могут располагаться:
- на кончиках пальцев;
- под ногтями;
- в зеве и гортани;
- в бронхах;
- в мочевыводящих путях и почечных лоханках;
- во влагалище у женщин.
Кровотечения меняют свою локализацию: возможно начало с одного носового хода, затем присоединение других (легочно-бронхиальных, кишечных, почечных). Интенсивность меняется от незначительной до упорных, плохо поддающихся лечению, длительностью по несколько дней и недель. Такие случаи приводят в анемизации пациента.
Классификация, опасность и лечение легочных АВМ
Легочные АВМ — редкая патология. На 15 000 последовательных вскрытий обнаружено только три случая легочных АВМ [6]. Клиника Майо (Рочестер, Миннесота, США, одна из крупнейших клиник в мире, в ней работает около 3800 врачей и ученых) сообщила о 194 случаях легочных АВМ в течение 45 лет — 4,3 случая в год [7, 8, 9]. Легочная АВМ может быть приобретенной или врожденной, и приблизительно 70 % случаев мальформаций связаны с болезнью Рандю — Ослера [10]. Легочные АВМ в два раза чаще встречаются у женщин, чем у мужчин [11].
Легочные АВМ могут быть простыми или сложными. Простой тип (80 % случаев) состоит из одной питающей артерии и одной отводящей вены, а сложный тип (20 % случаев) имеет две или более питающие артерии или отводящие вены [12, 13]. Более половины легочных АВМ находятся в нижних отделах, реже всего мальформации встречаются в верхних отделах [12, 14]. У пациентов с болезнью Рандю — Ослера легочные мальформации — множественные (35–65 %) [8, 15, 16] и обычно связаны с тяжелыми осложнениями и инфекциями [17]. У 25 % пациентов с синдромом Рандю — Ослера обнаружены двусторонние легочные АВМ [18].
У большинства пациентов с легочными АВМ имеется симптоматика, которая часто развивается между четвертым и шестым десятилетиями жизни, когда из артериальной системы в венозную через мальформации шунтируется более 25 % от общего объема циркулирующей крови [6, 19, 20]. Напомним, что в малом кругу кровообращения по артериям течет венозная кровь, а по венам — артериальная. Одышка при нагрузке является наиболее распространенной жалобой у пациентов с легочными АВМ, она наблюдается примерно у половины пациентов [15, 23, 24]. Другие симптомы: кровохарканье (примерно у 10 % пациентов), боль в груди (6 %), пальцы в форме барабанных палочек (20 %), цианоз (18 %) и торакальные шумы (3 %) [15]. Симптомы болезни Рандю — Ослера обычно становятся заметными до 20 лет (например, носовое кровотечение, телеангиоэктазии на коже) [21, 22]. У нашего пациента была одышка при нагрузке с хронической гипоксией, и он был устойчив к традиционной терапии астмы. Торакальный шум был обнаружен при физикальном осмотре.
Классический рентгенографический вид легочной АВМ — круглая или овальная масса однородной плотности, резко выраженная, чаще в нижних долях, диаметром от одного до пяти сантиметров. Чувствительность рентгенограммы грудной клетки составляет 70 % при диагностике АВМ [10]. У описанного пациента рентгенограмма грудной клетки показала однородную массу мягких тканей, которая могла оказаться опухолью. КТ-ангиография считается «золотым стандартом» для диагностики легочных АВМ с чувствительностью более 97 % [27].
Наличие легочных мальформаций чревато опасными для жизни осложнениями: инсульт, абсцесс головного мозга, гемоторакс и кровохарканье, особенно у женщин [28, 29]. Разрыв легочной мальформации может произойти в любом возрасте, независимо от размера поражения [30, 31, 32]. Без соответствующего лечения смертность болезни Рандю-Ослера с АВМ превышает 11 % [33].
Если все легочные мальформации размером более двух сантиметров, или если питающие артерии больше двух миллиметров в диаметре, следует лечить хирургически или с помощью эмболотерапии [8, 34].
По материалам: https://jmedicalcasereports.biomedcentral.com/articles/10.1186/1752-1947-7-32
Источники
- Churton T: Multiple aneurysms of the pulmonary artery. Br Med J. 1897, 1: 1223–1225.
- Global Initiative for Asthma (GINA), National Heart, Lung and Blood Institute (NHLBI): Global strategy for asthma management and prevention. 2006, Bethesda (MD): Global Initiative for Asthma (GINA), National Heart, Lung and Blood Institute (NHLBI), 339‑Available from: https://www.ginasthma.com
- Risenga SM: Difficult-to-control asthma in children — an overview. Curr Allergy Clin Immunol. 2011, 24: 18–21.
- Weiss P, Rundell KW: Imitators of exercise-induced bronchoconstriction. Allergy Asthma Clin Immunol. 2009, 5: 7–10.1186/1710‑1492‑5‑7.
- American Thoracic Society: Current understanding, recommendations, and unanswered questions. In Proceedings of the ATS workshop on refractory asthma. Am J Respir Crit Care Med. 2000, 162: 2341–2351.
- Sloan RD, Cooley RN: Congenital pulmonary arteriovenous aneurysm. Am J Roentgenol Radium Ther Nucl Med. 1953, 70: 183–210.
- Swanson KL, Prakash UB, Stanson AW: Pulmonary arteriovenous fistulas: Mayo Clinic experience, 1982–1997. Mayo Clin Proc. 1999, 74: 671–680. 10.4065/74.7.671.
- Dines DE, Arms RA, Bernatz PE, Gomes MR: Pulmonary arteriovenous fistulas. Mayo Clin Proc. 1974, 49: 460–465.
- Dines DE, Seward JB, Bernatz PE: Pulmonary arteriovenous fistulas. Mayo Clin Proc. 1983, 58: 176–181.
- Gossage JR, Kani G: Pulmonary arteriovenous malformation: a state of the art review. Am J Respir Crit Care Med. 1998, 158: 643–660.
- Van Gent MW, Post MC, Snijder RJ, Westermann CJ, Plokker HW, Mager JJ: Real prevalence of pulmonary right-to-left shunt according to genotype in patients with hereditary hemorrhagic telangiectasia: a transthoracic contrast echocardiography study. Chest. 2010, 138: 833–839. 10.1378/chest. 09–1849.
- Burke CM, Safai C, Nelson DP, Raffin TA: Pulmonary arteriovenous malformations: a critical update. Am Rev Respir Dis. 1986, 25: 331–334.
- Remy J, Remy-Jardin M, Wattinness L, Deffontaines C: Pulmonary AVMs — evaluation with CT of the chest before and after treatment. Radiology. 1992, 182: 809–816.
- Frazer RG, Pare JAP, Pare RD, Frazer RS, Genereux GP: Diagnosis of Diseases of the Chest. 1989, Philadelphia: B. Saunders Company, 3
- Cottin V, Chinet T, Lavole A, Corre R, Marchand E, Reynaud-Gaubert M, Plauchu H, Cordier JF: Groupe d’Etudes et de Recherche sur les Maladies ‘Orphelines’ Pulmonaires (GERM’O’P). Pulmonary arteriovenous malformations in hereditary hemorrhagic telangiectasia: a series of 126 patients. Medicine (Baltimore). 2007, 86: 1–17. 10.1097/MD. 0b013e31802f8da1.
- White RI, Lynch-Nyhan A, Terry P, Buescher PC, Farmlett EJ, Charnas L, Shuman K, Kim W, Kinnison M, Mitchell SE: Pulmonary arteriovenous malformations: techniques and long-term outcome of embolotherapy. Radiology. 1988, 169: 663–669.
- Hodgson CH, Burchel HB, Good CA, Clagett OT: Hereditary hemorrhagic telangiectasia and pulmonary arteriovenous fistula. Survey of a large family. N EngI J Med. 1959, 261: 625–636. 10.1056/NEJM195909242611301.
- Bosher LH, Blake DA, Byrd BR: An analysis of the pathologic anatomy of pulmonary arteriovenous aneurysms with particular reference to the applicability of local excision. Surgery. 1959, 45: 91–104.
- Fuchizaki U, Miyamori H, Kitagawa S, Kaneko S, Kobayashi K: Hereditary haemorrhagic telangiectasia (Rendu — Osler — Weber disease). Lancet. 2003, 362: 1490–1494. 10.1016/S0140–6736 (03) 14696‑X.
- Chauhan MS, Ahuja JM, Tewari SC, Jayaswal R, Dandona PK: Osler — Rendu — Weber disease presenting as recurrent hemoptysis. Indian J Chest Dis Allied Sci. 1989, 31: 227–232.
- Vase P, Holm M, Arendrup H: Pulmonary arteriovenous fistulas in hereditary hemorrhagic telangiectasia. Acta Med Scand. 1985, 218: 105–109.
- Plauchu H, de Chadarévian JP, Bideau A, Robert JM: Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet. 1989, 32: 291–297. 10.1002/ajmg. 1320320302.
- Begbie ME, Wallace GM, Shovlin CL: Hereditary haemorrhagic telangiectasia (Osler — Weber — Rendu syndrome): a view from the 21st century. Postgrad Med J. 2003, 79: 18–24. 10.1136/pmj. 79.927.18.
- Shovlin CL, Letarte M: Hereditary haemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax. 1999, 54: 714–729. 10.1136/thx. 54.8.714.
- Dutton JA, Jackson JE, Hughes JM, Whyte MK, Peters AM, Ussov W, Allison DJ: Pulmonary arteriovenous malformations: results of treatment with coil embolization in 53 patients. AJR Am J Roentgenol. 1995, 165: 1119–1125.
- Gupta P, Mordin C, Curtis J, Hughes JM, Shovlin CL, Jackson JE: Pulmonary arteriovenous malformations: effect of embolization on right-to-left shunt, hypoxemia, and exercise tolerance in 66 patients. AJR Am J Roentgeno. 2002, 179: 347–355.
- Remy J, Remy-Jardin M, Giraud F, Wattinne L: Angioarchitecture of pulmonary arteriovenous malformations: clinical utility of three-dimensional helical CT. Radiology. 1994, 191: 657–664.
- Dinkel HP, Triller J: Pulmonary arteriovenous malformations: embolotherapy with superselective coaxial catheter placement and filling of venous sac with Guglielmi detachable coils. Radiology. 2002, 223: 709–714. 10.1148/radiol. 2233010953.
- Moore BP: Pulmonary arteriovenous fistula [abstract]. Thorax. 1969, 24: s381 —
- Bowers WF: Rupture of visceral hemangioma as cause of death with report of a case of pulmonary hemangioma. Nebr Med J. 1936, 21: 55–57.
- Shashy SS, Jones BC, Kitchens CS: Spontaneous hemothorax in a patient with Osler — Weber — Rendu disease. South Med J. 1985, 78: 1393–1394. 10.1097/00007611‑198511000‑00039.
- Chanatry BJ: Acute hemothorax owing to pulmonary arteriovenous malformation in pregnancy. Anesth Anaig. 1992, 74: 613–615.
- Najarian KE, Morris CS: Arterial embolization in the chest. J Thorac Imaging. 1998, 13: 93–104. 10.1097/00005382‑199804000‑00004.
- Faughnan ME, Palda VA, Garcia-Tsao G, Geisthoff UW, McDonald J, Proctor DD, Spears J, Brown DH, Buscarini E, Chesnutt MS, Cottin V, Ganguly A, Gossage JR, Guttmacher AE, Hyland RH, Kennedy SJ, Korzenik J, Mager JJ, Ozanne AP, Piccirillo JF, Picus D, Plauchu H, Porteous ME, Pyeritz RE, Ross DA, Sabba C, Swanson K, Terry P, Wallace MC, Westermann CJ, White RI, Young LH, Zarrabeitia R, HHT Foundation International — Guidelines Working Group: International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. 2011, 48: 73–87. 10.1136/jmg. 2009.069013.
Как проявляется неполноценность сосудов во внутренних органах?
Врожденные нарушения строения стенки мелких сосудов образуют характерные артериовенозные аневризмы. Чаще всего они находятся в легочной ткани. К кожным симптомам добавляются:
Еще советуем:Лазерная коагуляция телеангиоэктазий
- одышка;
- инъецированность склер;
- синюшно-красный цвет лица;
- полиглобулия — увеличение количества эритроцитов.
Если аневризматические расширения капиллярного русла появляются в печени, почках или селезенке, их очень сложно диагностировать. Чаще всего заболевание принимается за опухоли, туберкулез внутренних органов, эритремию, врожденные пороки.
Длительное течение ангиоматоза приводит к:
- прогрессирующей легочно-сердечной недостаточности;
- хронической уремии;
- тяжелой постгеморрагической анемии;
- сердечной недостаточности.
Изменения необратимы.
Как развивается
Так как заболевание происходит из-за генетической предрасположенности его можно диагностировать с ранних лет жизни ребенка. Вероятные внутриутробные отклонения у малыша можно заметить по состоянию матери во время беременности.
Патология встречается крайне редко, меньше, чем у 0.1% населения. Болезнь характеризуется следующими факторами:
- гипохромной анемией;
- стойким расширением мелких сосудов;
- венозными и капиллярными сетками на теле;
- синдромом Луи-Бара.
Большинство первичных симптомов обусловлено эмбриональным нарушением роста плода в третьем семестре. Оно влечет за собой хроническое и генетически передаваемое заболевание.
Особенности клинического течения
Течение болезни Рандю-Ослера некоторыми учеными делится на стадии, которые носят аналогичные названия с формами по Ослеру. Они придерживаются мнения, что по форме высыпаний можно судить о выраженности патологических изменений в сосудах.
Выделяют типы патологии:
- назальный — проявляется носовыми кровотечениями;
- глоточный — телеангиэктазии видны при осмотре глотки;
- кожный — кровоточат отдельные участки на коже;
- висцеральный — отличается кровоточивостью внутренних органов;
- смешанный — поражены и кожа, и слизистые внутренних органов.
Разновидность кожной локализации — пятна возникают на пальцах и ладонной поверхности
Причины возникновения
Причиной возникновения синдрома Ослера является мутация двух генов, которые отвечают за процессы восстановления тканей и морфогенез (формообразование) сосудов.
Передается синдром Рандю — Ослера — Вебера по аутосомно-доминантному принципу: развитие болезни у ребенка происходит при передаче поврежденного гена от любого из родителей. Ненаследственные патологии фиксируются крайне редко. Считается, что причиной изменения структуры стенок сосудов являются неблагоприятные факторы (чаще всего это химические вещества и инфекции), которым подвергается плод на протяжении беременности.
Первые симптомы болезни Рандю — Ослера можно наблюдать у детей 6-10 лет. Как правило, они проявляются на голове, мочках ушей, слизистых десен, щек, губ и на крыльях носа. По мере взросления количество ангиэктазий увеличивается, а кровоточат они чаще и сильнее.
Диагностика
При наличии типичных поверхностных изменений на лице, видимых слизистых, волосистой части головы, при повторных кровотечениях диагноз не вызывает сложности.
По анализам крови невозможно судить о болезни Рандю-Ослера, поскольку не выявляются существенные изменения гемостаза. Имеются лишь последствия кровотечений, кровопотери в виде:
- умеренного тромбоцитоза,
- склонности к гиперкоагуляции,
- анемии,
- эритроцитоза,
- снижения гемоглобина.
При множественных телеангиэктазиях в крови наблюдается рост внутрисосудистого свертывания (коагулопатия потребления), тромбоцитопения.
Биохимические тесты помогают выявить повреждение органов и нарушение их функции.
В анализе мочи возможно обнаружение эритроцитов (гематурия). Массивное кровотечение, возможно, связано с почечнокаменной болезнью, распадающейся опухолью. Микрогематурия и белок больше указывают на гломерулонефрит.
Кровь в каловых массах указывает на кровотечение из желудка или кишечника. Для выявления точной локализации необходимы исследования с помощью эндоскопической техники. Оптические приборы позволяют увидеть типичное поражение слизистой.
Компьютерная томография проводится в случаях висцеральных форм.
Как лечат кровотечения при болезни Рандю-Ослера?
Лечение проводится, по сути, не самой болезни, а кровотечений.
Схематично алгоритм остановки кровотечения выглядит так:
- если источник кровотечения в доступном месте, использовать прижатие сосуда, гемостатическую губку, тампонаду с перекисью водорода, в лечебном учреждении — коагуляцию специальным прибором;
- при недоступном подозреваемом источнике препаратами выбора могут быть кислота аминокапроновая, Менадион (1% раствор), хлористый кальций (10% раствор), переливание крови и заменителей при тяжелых анемиях.
Более подробно консервативная терапия кровотечения заключается в использовании средств местного и общего воздействия.
Способ орошения врач-специалист применяет курсами
К местным относят:
- Орошения кровоточащей слизистой препаратами Лебетоксом, Рептилазой, Стипвеном, тромбином, тромбопластином, охлажденным раствором аминокапроновой кислоты. Специалисты рекомендуют постоянно держать дома в холодильнике одно из этих средств. Они могут понадобиться для оказания доврачебной помощи при кровотечении. Орошение проводят маленькой резиновой грушей или шприцем. Вставлять тампон не рекомендуется, поскольку он вызывает дополнительное травмирование слизистой.
- Для прижиганий используются растворы трихлоруксусной или хромовой кислот, нитрата серебра или применяют метод диатермокоагуляции.
При повторных профузных кровотечениях в ЛОР-отделениях производят:
- хирургическую отслойку слизистой с перевязкой приводящей артерии (решетчатой и верхнечелюстной);
- криоаппликацию — процедура заключается во введении в носовые ходы специального аппарата с циркулирующим азотом, который обеспечивает замораживание тканей до минус 196 градусов по Цельсию, воздействие проводится в течение 30–90 секунд.
Для закрепления эффекта дополнительно через день еще делают от 4 до 8 процедур секундного распыления жидкого азота. Результативность метода позволяет избавиться от носовых кровотечений на срок от нескольких месяцев до года.
После остановки носового кровотечения рекомендуется применять местно для регенерации и смягчения эпителия мази с ланолином, витамином Е, Декспантенолом
Для общего воздействия оказались полезными инъекции эстрогенов или тестостерона, половые гормоны смягчают кровоточивость. А такие известные препараты, как Викасол, Желатин, Дицинон, кальция хлорид, аминокапроновая кислота, при болезни Рандю-Ослера бесполезны.
Хирургическое лечение при висцеральных кровотечениях дает только временный эффект, поскольку вырастают новые ангиэктазии, и кровотечение возобновляется. Тем не менее оперировать стараются на ранних стадиях, пока в органах не наступили выраженные расстройства микроциркуляции.
При значительной кровопотере используют препараты из донорской крови (замороженную плазму и тромбоциты). Тяжелая форма анемии требует трансфузии эритроцитарной массы. Для полноценного лечения анемии пациентам назначают: препараты железа, витамин С.
Как протекает беременность у женщин с болезнью Рандю-Ослера?
Акушеры-гинекологи относят беременных женщин с болезнью Рандю-Ослера к группе высокого риска, они ведутся совместно с гематологом. Осложнений ждут во втором и третьем триместрах. Женщина должна быть предупреждена и заранее госпитализирована.
Все обследования с помощью эндоскопических аппаратов должны проводиться до наступления беременности, поскольку женщине в положении они уже противопоказаны. Это касается и магниторезонансной томографии.
Серьезные осложнения случаются редко. Роды проходят без внешних усилий и ведутся акушерами так же, как обычно. Кровотечения случаются очень редко. Возможно, женщину защищают гормональные изменения.
Прогноз заболевания
Прогноз болезни Рандю-Ослера зависит от того, насколько выражены сосудистые нарушения, от их локализации и семейных особенностей. Ухудшает прогноз преобладание церебральных симптомов, которые способны привести к эпилепсии, инсульту и абсцессам мозга. Без лечения любая форма синдрома Рандю-Ослера грозит тяжелыми осложнениями: массивные кровотечения и анемия, слепота после кровоизлияния в сетчатку глаза, легочная гипертензия; сердечная и другие виды недостаточности; цирроз печени.
Семейная геморрагическая телеангиэктазия – редкое заболевание, и его проявления совпадают с признаками многих болезненных состояний, например с патологиями печени. Поэтому при повышенной кровоточивости сосудов, беспричинных кровотечениях, появлении сосудистых рисунков на коже или анемии не затягивайте с визитом к врачу, а после постановки диагноза регулярно проходите обследования.