Rendu-Osler disease belongs to hereditary vasopathies, more precisely, it is called disseminated hemangiomatosis. The thinning of capillary walls by the age of six causes a change in the structure and turns them into small bleeding aneurysms, which disrupts local blood circulation in the tissues.
The first data about the clinic were described by the French physician Louis Marie Randu, and later more complete information was provided by Sir William Osler and Frederick Parks Weber at the end of the 19th and beginning of the 20th centuries. Therefore, the full name of the pathology is Rendu-Osler-Weber disease.
It is detected in 1 person out of 5000 people. Both boys and girls get sick equally often. It manifests itself to its maximum in middle age (40–50 years). During treatment, one has to deal with repeated bleeding that is difficult to stop.
In ICD-10, pathology is included in the subclass “Diseases of the capillaries” with code I78.0.
What is known about the causes of the disease?
To date, the exact cause of the disease has not been clarified. A connection has been established with genetic mutations and the inheritance of altered genes according to the dominant type, if present in one of the parents. However, there are cases independent of inheritance; they are called sporadic.
Genetic studies have made it possible to identify 3 types of disorders:
- Type I - the gene responsible for collagen synthesis;
- Type II—defect of tumor growth factor receptors;
- Type III - has complex markings (600604, 12p11–p12; Â).
As a result, the main sources of bleeding are:
- foci of thinning of the walls of capillaries of the venous and arterial type;
- uncontrolled expansion of the diameter of microvessels;
- arteriovenous aneurysms.
The negative impact of infectious diseases suffered by the expectant mother in the first trimester of pregnancy and taking medications has been established.
Provoking factors for bleeding can be:
- lack of fortification of food, vegetarian diet;
- tight-fitting clothing that injures superficial vessels.
Modern ideas about the disease
Modern data have made it possible to establish the basis for the violation of the anatomical integrity of blood vessels (dysplasia). Inferiority of the mesenchymal membrane leads to the partial absence of elastic and muscle fibers and their replacement with loose connective tissue.
In areas of venules and arterioles, protrusions such as aneurysmal dilatations (telangiectasis) are formed.
Minor injury causes bleeding. Damage to capillaries in Randu-Osler disease has not only a superficial skin localization, but spreads to the mucous membranes of internal organs, most often the bronchi, nasopharynx, oral cavity, and bladder.
Treatment effectiveness
Therapy is prescribed both taking into account anamnestic data and based on the results of the examination. The tactics to combat pathology are determined by the doctor. The basis of treatment for Randu-Osler disease is the use of medications aimed at stopping existing bleeding and preventing the occurrence of new ones. For this purpose, drugs such as Vikasol, Etamzilate and Aminocaproic acid are prescribed. They are used both in injection form and for local treatment of affected areas. These substances help normalize the rheological properties of blood.
If a patient has defects in large vessels, such as an aortic aneurysm, surgical intervention is warranted. Laser techniques and cryodestruction are used to treat minor lesions.
In rare cases, patients require hospitalization. It is justified in cases of significant bleeding. After restoration of the integrity of the vessels, patients are treated in the intensive care unit. Fluid loss is corrected through infusions. In some cases, blood transfusion is required.
Clinical manifestations
Despite hereditary transmission, Randu-Osler disease begins to manifest itself after 10 years, and more often during puberty and by the age of 40.
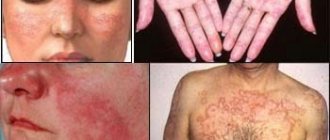
The first signs are expressed by vascular “stars” with dark red papules in the center. They are located:
- on the skin of the nose,
- on the lips,
- in the mouth (gums, tongue),
- on the ears,
- in the area of the scalp.
When injured, angioectasis bleeds. The only possible symptom is nosebleeds.
With age, the number of rashes increases, they spread to the mucous membranes of different organs. The stomach and intestines are most affected. Therefore, bleeding from the gastrointestinal tract is added to the symptoms.
When similar formations in the lining of the brain rupture, the clinical picture of a stroke appears. Girls and women may experience uterine bleeding and heavy menstruation.
Repeated bleeding is accompanied by the development of iron deficiency anemia. Patients develop:
- weakness,
- headache,
- dizziness.
The intermediate form of telangiectasias is distinguished by vascular packs around the spot in the center
Osler identified 3 types of telangiectasia:
- early ones - have the form of small spots with irregular outlines;
- intermediate - characterized by vascular processes (anastomoses between arterioles and venules);
- nodular - bright red-bluish nodules are formed in the center of the rash, protruding above the skin by 1-3 mm, their diameter reaches 5-7 mm.
Patients over 25 years of age have two or all types of angiectasias. To distinguish them from other vascular formations, you need to lightly press on the surface, the spot will turn pale and fill with blood again after contact stops.
Telangiectasia can be located:
- at the tips of the fingers;
- under the nails;
- in the pharynx and larynx;
- in the bronchi;
- in the urinary tract and renal pelvis;
- in the vagina in women.
Bleeding changes its localization: it may begin with one nasal passage, then the addition of others (pulmonary-bronchial, intestinal, renal). The intensity varies from insignificant to persistent, difficult to treat, lasting several days and weeks. Such cases lead to anemia in the patient.
Classification, danger and treatment of pulmonary AVMs
Pulmonary AVMs are a rare pathology. In 15,000 consecutive autopsies, only three cases of pulmonary AVMs were found [6]. The Mayo Clinic (Rochester, Minnesota, USA, one of the largest hospitals in the world, with approximately 3800 physicians and scientists) reported 194 cases of pulmonary AVMs over 45 years—4.3 cases per year [7, 8, 9]. Pulmonary AVM can be acquired or congenital, and approximately 70% of malformations are associated with Randu-Osler disease [10]. Pulmonary AVMs are twice as common in women as in men [11].
Pulmonary AVMs can be simple or complex. The simple type (80% of cases) consists of one feeding artery and one draining vein, and the complex type (20% of cases) has two or more feeding arteries or draining veins [12, 13]. More than half of pulmonary AVMs are located in the lower sections; malformations are least common in the upper sections [12, 14]. In patients with Rendu-Osler disease, pulmonary malformations are multiple (35–65%) [8, 15, 16] and are usually associated with severe complications and infections [17]. Bilateral pulmonary AVMs were found in 25% of patients with Randu-Osler syndrome [18].
Most patients with pulmonary AVMs have symptoms, which often develop between the fourth and sixth decades of life, when more than 25% of the total circulating blood volume is shunted from the arterial system to the venous system through the malformations [6, 19, 20]. Let us recall that in the pulmonary circulation venous blood flows through the arteries, and arterial blood flows through the veins. Dyspnea on exertion is the most common complaint in patients with pulmonary AVMs, occurring in approximately half of patients [15, 23, 24]. Other symptoms include hemoptysis (approximately 10% of patients), chest pain (6%), drumstick fingers (20%), cyanosis (18%), and thoracic murmurs (3%) [15]. Symptoms of Randu-Osler disease usually become noticeable before age 20 (eg, epistaxis, skin telangiectasias) [21, 22]. Our patient had exertional dyspnea with chronic hypoxia and was resistant to conventional asthma therapy. A thoracic murmur was detected on physical examination.
The classic radiographic appearance of a pulmonary AVM is a round or oval mass of uniform density, pronounced, often in the lower lobes, with a diameter of one to five centimeters. The sensitivity of chest X-ray is 70% in diagnosing AVM [10]. In the patient described, a chest x-ray showed a homogeneous soft tissue mass that may have been a tumor. CT angiography is considered the gold standard for diagnosing pulmonary AVMs with a sensitivity of more than 97% [27].
The presence of pulmonary malformations is fraught with life-threatening complications: stroke, brain abscess, hemothorax and hemoptysis, especially in women [28, 29]. Rupture of a pulmonary malformation can occur at any age, regardless of the size of the lesion [30, 31, 32]. Without appropriate treatment, the mortality rate of Rendu-Osler disease with AVM exceeds 11% [33].
If all pulmonary malformations are larger than two centimeters in size, or if the feeding arteries are larger than two millimeters in diameter, they should be treated surgically or with embolotherapy [8, 34].
Based on materials from: https://jmedicalcasereports.biomedcentral.com/articles/10.1186/1752-1947-7-32
Sources
- Churton T: Multiple aneurysms of the pulmonary artery. Br Med J 1897, 1:1223–1225.
- Global Initiative for Asthma (GINA), National Heart, Lung and Blood Institute (NHLBI): Global strategy for asthma management and prevention. 2006, Bethesda (MD): Global Initiative for Asthma (GINA), National Heart, Lung and Blood Institute (NHLBI), 339‑Available from: https://www.ginasthma.com
- Risenga SM: Difficult-to-control asthma in children - an overview. Curr Allergy Clin Immunol. 2011, 24: 18–21.
- Weiss P, Rundell KW: Imitators of exercise-induced bronchoconstriction. Allergy Asthma Clin Immunol. 2009, 5: 7–10.1186/1710‑1492‑5‑7.
- American Thoracic Society: Current understanding, recommendations, and unanswered questions. In Proceedings of the ATS workshop on refractory asthma. Am J Respir Crit Care Med. 2000, 162: 2341–2351.
- Sloan RD, Cooley RN: Congenital pulmonary arteriovenous aneurysm. Am J Roentgenol Radium Ther Nucl Med. 1953, 70: 183–210.
- Swanson KL, Prakash UB, Stanson AW: Pulmonary arteriovenous fistulas: Mayo Clinic experience, 1982–1997. Mayo Clin Proc. 1999, 74: 671–680. 10.4065/74.7.671.
- Dines DE, Arms RA, Bernatz PE, Gomes MR: Pulmonary arteriovenous fistulas. Mayo Clin Proc. 1974, 49: 460–465.
- Dines DE, Seward JB, Bernatz PE: Pulmonary arteriovenous fistulas. Mayo Clin Proc. 1983, 58: 176–181.
- Gossage JR, Kani G: Pulmonary arteriovenous malformation: a state of the art review. Am J Respir Crit Care Med. 1998, 158: 643–660.
- Van Gent MW, Post MC, Snijder RJ, Westermann CJ, Plokker HW, Mager JJ: Real prevalence of pulmonary right-to-left shunt according to genotype in patients with hereditary hemorrhagic telangiectasia: a transthoracic contrast echocardiography study. Chest. 2010, 138: 833–839. 10.1378/chest. 09–1849.
- Burke CM, Safai C, Nelson DP, Raffin TA: Pulmonary arteriovenous malformations: a critical update. Am Rev Respir Dis. 1986, 25: 331–334.
- Remy J, Remy-Jardin M, Wattinness L, Deffontaines C: Pulmonary AVMs—evaluation with CT of the chest before and after treatment. Radiology. 1992, 182: 809–816.
- Frazer RG, Pare JAP, Pare RD, Frazer RS, Genereux GP: Diagnosis of Diseases of the Chest. 1989, Philadelphia: B. Saunders Company, 3
- Cottin V, Chinet T, Lavole A, Corre R, Marchand E, Reynaud-Gaubert M, Plauchu H, Cordier JF: Groupe d'Etudes et de Recherche sur les Maladies 'Orphelines' Pulmonaires (GERM'O'P). Pulmonary arteriovenous malformations in hereditary hemorrhagic telangiectasia: a series of 126 patients. Medicine (Baltimore). 2007, 86: 1–17. 10.1097/MD. 0b013e31802f8da1.
- White RI, Lynch-Nyhan A, Terry P, Buescher PC, Farmlett EJ, Charnas L, Shuman K, Kim W, Kinnison M, Mitchell SE: Pulmonary arteriovenous malformations: techniques and long-term outcome of embolotherapy. Radiology. 1988, 169: 663–669.
- Hodgson CH, Burchel HB, Good CA, Clagett OT: Hereditary hemorrhagic telangiectasia and pulmonary arteriovenous fistula. Survey of a large family. N Eng I J Med. 1959, 261: 625–636. 10.1056/NEJM195909242611301.
- Bosher LH, Blake DA, Byrd BR: An analysis of the pathologic anatomy of pulmonary arteriovenous aneurysms with particular reference to the applicability of local excision. Surgery. 1959, 45: 91–104.
- Fuchizaki U, Miyamori H, Kitagawa S, Kaneko S, Kobayashi K: Hereditary haemorrhagic telangiectasia (Rendu-Osler-Weber disease). Lancet. 2003, 362: 1490–1494. 10.1016/S0140–6736 (03) 14696‑X.
- Chauhan MS, Ahuja JM, Tewari SC, Jayaswal R, Dandona PK: Osler - Rendu - Weber disease presenting as recurrent hemoptysis. Indian J Chest Dis Allied Sci. 1989, 31: 227–232.
- Vase P, Holm M, Arendrup H: Pulmonary arteriovenous fistulas in hereditary hemorrhagic telangiectasia. Acta Med Scand. 1985, 218: 105–109.
- Plauchu H, de Chadarévian JP, Bideau A, Robert JM: Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet. 1989, 32: 291–297. 10.1002/ajmg. 1320320302.
- Begbie ME, Wallace GM, Shovlin CL: Hereditary haemorrhagic telangiectasia (Osler - Weber - Rendu syndrome): a view from the 21st century. Postgrad Med J 2003, 79:18–24. 10.1136/pmj. 79.927.18.
- Shovlin CL, Letarte M: Hereditary haemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax. 1999, 54: 714–729. 10.1136/thx. 54.8.714.
- Dutton JA, Jackson JE, Hughes JM, Whyte MK, Peters AM, Ussov W, Allison DJ: Pulmonary arteriovenous malformations: results of treatment with coil embolization in 53 patients. AJR Am J Roentgenol. 1995, 165: 1119–1125.
- Gupta P, Mordin C, Curtis J, Hughes JM, Shovlin CL, Jackson JE: Pulmonary arteriovenous malformations: effect of embolization on right-to-left shunt, hypoxemia, and exercise tolerance in 66 patients. AJR Am J Roentgeno. 2002, 179: 347–355.
- Remy J, Remy-Jardin M, Giraud F, Wattinne L: Angioarchitecture of pulmonary arteriovenous malformations: clinical utility of three-dimensional helical CT. Radiology. 1994, 191: 657–664.
- Dinkel HP, Triller J: Pulmonary arteriovenous malformations: embolotherapy with superselective coaxial catheter placement and filling of venous sac with Guglielmi detachable coils. Radiology. 2002, 223: 709–714. 10.1148/radiol. 2233010953.
- Moore BP: Pulmonary arteriovenous fistula [abstract]. Thorax. 1969, 24: s381 -
- Bowers WF: Rupture of visceral hemangioma as cause of death with report of a case of pulmonary hemangioma. Nebr Med J 1936, 21:55–57.
- Shashy SS, Jones BC, Kitchens CS: Spontaneous hemothorax in a patient with Osler - Weber - Rendu disease. South Med J 1985, 78:1393–1394. 10.1097/00007611‑198511000‑00039.
- Chanatry BJ: Acute hemothorax owing to pulmonary arteriovenous malformation in pregnancy. Anesth Anaig. 1992, 74: 613–615.
- Najarian KE, Morris CS: Arterial embolization in the chest. J Thorac Imaging. 1998, 13: 93–104. 10.1097/00005382‑199804000‑00004.
- Faughnan ME, Palda VA, Garcia-Tsao G, Geisthoff UW, McDonald J, Proctor DD, Spears J, Brown DH, Buscarini E, Chesnutt MS, Cottin V, Ganguly A, Gossage JR, Guttmacher AE, Hyland RH, Kennedy SJ, Korzenik J, Mager JJ, Ozanne AP, Piccirillo JF, Picus D, Plauchu H, Porteous ME, Pyeritz RE, Ross DA, Sabba C, Swanson K, Terry P, Wallace MC, Westermann CJ, White RI, Young LH, Zarrabeitia R , HHT Foundation International - Guidelines Working Group: International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. 2011, 48: 73–87. 10.1136/jmg. 2009.069013.
How does vascular deficiency manifest itself in internal organs?
Congenital defects in the structure of the walls of small vessels form characteristic arteriovenous aneurysms. Most often they are found in the lung tissue. Skin symptoms include:
We also recommend: Laser coagulation of telangiectasia
- dyspnea;
- scleral injection;
- bluish-red complexion;
- polyglobulia - an increase in the number of red blood cells.
If aneurysmal dilatations of the capillary bed appear in the liver, kidneys or spleen, they are very difficult to diagnose. Most often, the disease is mistaken for tumors, tuberculosis of internal organs, erythremia, and congenital defects.
Long-term course of angiomatosis leads to:
- progressive pulmonary heart failure;
- chronic uremia;
- severe posthemorrhagic anemia;
- heart failure.
The changes are irreversible.
How it develops
Since the disease occurs due to genetic predisposition, it can be diagnosed from the early years of a child’s life. Possible intrauterine abnormalities in the baby can be noticed by the condition of the mother during pregnancy.
The pathology is extremely rare, less than 0.1% of the population. The disease is characterized by the following factors:
- hypochromic anemia;
- persistent dilation of small vessels;
- venous and capillary networks on the body;
- Louis-Bar syndrome.
Most of the primary symptoms are caused by embryonic growth disorder of the fetus in the third semester. It entails a chronic and genetically transmitted disease.
Features of the clinical course
The course of Randu-Osler disease is divided by some scientists into stages, which have similar names to the Osler forms. They are of the opinion that the shape of the rash can be used to judge the severity of pathological changes in the blood vessels.
There are types of pathology:
- nasal - manifested by nosebleeds;
- pharyngeal - telangiectasias are visible when examining the pharynx;
- cutaneous - individual areas of the skin bleed;
- visceral - characterized by bleeding of internal organs;
- mixed - both the skin and mucous membranes of internal organs are affected.
A type of skin localization - spots appear on the fingers and palmar surface
Causes
The cause of Osler syndrome is a mutation of two genes that are responsible for the processes of tissue repair and morphogenesis (shape formation) of blood vessels.
Randu-Osler-Weber syndrome is transmitted according to an autosomal dominant principle: the development of the disease in a child occurs when a damaged gene is transmitted from either parent. Non-hereditary pathologies are recorded extremely rarely. It is believed that the cause of changes in the structure of the walls of blood vessels are unfavorable factors (most often chemicals and infections) to which the fetus is exposed during pregnancy.
The first symptoms of Randu-Osler disease can be observed in children 6-10 years old. As a rule, they appear on the head, earlobes, mucous membranes of the gums, cheeks, lips and on the wings of the nose. As you get older, the number of angiectasias increases, and they bleed more often and more severely.
Diagnostics
In the presence of typical superficial changes on the face, visible mucous membranes, scalp, and repeated bleeding, the diagnosis is not difficult.
It is impossible to judge Rendu-Osler disease from blood tests, since significant changes in hemostasis are not detected. There are only consequences of bleeding, blood loss in the form of:
- moderate thrombocytosis,
- tendency to hypercoagulability,
- anemia,
- erythrocytosis,
- decrease in hemoglobin.
With multiple telangiectasias in the blood, an increase in intravascular coagulation (consumption coagulopathy) and thrombocytopenia are observed.
Biochemical tests help identify organ damage and dysfunction.
A urine test may detect red blood cells (hematuria). Massive bleeding may be due to kidney stones, a disintegrating tumor. Microhematuria and protein are more suggestive of glomerulonephritis.
Blood in the stool indicates bleeding from the stomach or intestines. To identify the exact localization, studies using endoscopic techniques are necessary. Optical instruments allow you to see typical mucosal lesions.
Computed tomography is performed in cases of visceral forms.
How is bleeding in Randu-Osler disease treated?
The treatment is, in fact, not the disease itself, but the bleeding.
Schematically, the algorithm for stopping bleeding looks like this:
- if the source of bleeding is in an accessible place, use pressure on the vessel, a hemostatic sponge, tamponade with hydrogen peroxide, or in a medical institution - coagulation with a special device;
- if the suspected source is inaccessible, the drugs of choice may be aminocaproic acid, Menadione (1% solution), calcium chloride (10% solution), blood transfusions and blood substitutes for severe anemia.
In more detail, conservative therapy for bleeding involves the use of local and general agents.
The irrigation method is applied by a specialist doctor in courses
Locals include:
- Irrigation of bleeding mucosa with Lebetox, Reptilase, Stipven, thrombin, thromboplastin, cooled solution of aminocaproic acid. Experts recommend keeping one of these products in the refrigerator at home at all times. They may be needed to provide first aid for bleeding. Irrigation is carried out with a small rubber bulb or syringe. Inserting a tampon is not recommended, as it causes additional trauma to the mucous membrane.
- For cauterization, solutions of trichloroacetic or chromic acids, silver nitrate are used, or the diathermocoagulation method is used.
In case of repeated profuse bleeding in ENT departments the following is performed:
- surgical detachment of the mucous membrane with ligation of the adductor artery (ethmoidal and maxillary);
- cryoapplication - the procedure involves introducing a special device with circulating nitrogen into the nasal passages, which ensures tissue freezing to minus 196 degrees Celsius, the effect is carried out for 30-90 seconds.
To consolidate the effect, an additional 4 to 8 procedures of one-second spraying of liquid nitrogen are done every other day. The effectiveness of the method allows you to get rid of nosebleeds for a period of several months to a year.
After stopping nosebleeds, it is recommended to apply ointments with lanolin, vitamin E, Dexpanthenol topically to regenerate and soften the epithelium
For general effects, injections of estrogen or testosterone have proven useful; sex hormones soften bleeding. And such well-known drugs as Vikasol, Gelatin, Dicynone, calcium chloride, aminocaproic acid are useless for Rendu-Osler disease.
Surgical treatment for visceral bleeding gives only a temporary effect, since new angiectasia grows and bleeding resumes. Nevertheless, they try to operate in the early stages, before pronounced microcirculation disorders occur in the organs.
In case of significant blood loss, drugs from donor blood (frozen plasma and platelets) are used. Severe anemia requires red blood cell transfusion. For complete treatment of anemia, patients are prescribed: iron supplements, vitamin C.
How does pregnancy proceed in women with Rendu-Osler disease?
Obstetricians-gynecologists classify pregnant women with Rendu-Osler disease as a high-risk group; they are managed together with a hematologist. Complications are expected in the second and third trimesters. The woman should be warned and hospitalized in advance.
All examinations using endoscopic devices should be carried out before pregnancy, since they are already contraindicated for pregnant women. This also applies to magnetic resonance imaging.
Serious complications are rare. The birth takes place without external forces and is carried out by obstetricians in the same way as usual. Bleeding occurs very rarely. Perhaps the woman is protected by hormonal changes.
Disease prognosis
The prognosis of Randu-Osler disease depends on how severe the vascular disorders are, their location and family characteristics. The prognosis is worsened by the predominance of cerebral symptoms, which can lead to epilepsy, stroke and brain abscesses. Without treatment, any form of Randu-Osler syndrome can lead to severe complications: massive bleeding and anemia, blindness after retinal hemorrhage, pulmonary hypertension; heart and other types of failure; cirrhosis of the liver.
Familial hemorrhagic telangiectasia is a rare disease, and its manifestations coincide with those of many disease states, such as liver pathologies. Therefore, if there is increased bleeding of blood vessels, unreasonable bleeding, the appearance of vascular patterns on the skin or anemia, do not delay a visit to the doctor, and after making a diagnosis, undergo regular examinations.