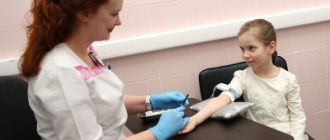
Platelets are unique blood cells that provide one of the main processes of human life support - blood clotting. Thrombocytopathy is a collective name for diseases that are united by one aspect: the quality of the platelets themselves is impaired, but their quantity remains within the age norm. The pathology is quite common, although it can manifest itself in varying degrees of severity. Since etiology and pathogenesis manifest themselves, what are the principles of treatment and the main symptoms of the disease?
Thrombocytopathy
Classification of thrombocytopathies
Thrombocytopathy is divided into types: primary and secondary. The differences between them are as follows: primary occurs as a hereditary pathology and manifests itself immediately after birth, and secondary thrombocytopathy is diagnosed as a complication of somatic diseases. When systematizing pathology, the diagnosis “Disaggregation thrombocytopathy” is separately identified, which is characterized by syndromes of hemorrhagic and post-hemorrhagic diathesis. Moreover, this disease is more typical for girls and is detected during a number of laboratory tests at an early age. Among the hereditary forms, the following types of thrombocytopathies are distinguished:
- Associated with membrane abnormality. These include Bernard-Soulier syndrome, pseudovon Willebrand disease, Glanzmann thrombasthenia, etc.
- Associated with intracellular abnormalities and impaired platelet aggregation, the pathogenesis of such forms of pathology is due to a deficiency of α-granules or a violation of their release from cells. This group includes Hermansky-Pudlak disease, gray platelet syndrome, Chediak-Higashi syndrome, etc.
- Platelet dysfunction in vascular dysplasias (for example, Ehlers-Danlos disease).
Mixed platelet disorders are also classified as a separate group. These are May-Hegglin, Wiskott-Aldrich syndromes, etc. Often such pathologies are accompanied by other congenital anomalies, for example, Down syndrome. They are usually diagnosed during screening at the end of the first trimester of pregnancy.
Treatment
Patients with any form of thrombocytopathy are required to be registered at a dispensary under the supervision of a hematologist (periodically examining the state of hemostasis).
The first step to treatment is to eliminate the negative effects of adverse factors that increase bleeding. For example, alcohol and certain dishes (homemade preparations with vinegar and salicylates) are strictly prohibited. But vitamins (A, P, C), on the contrary, occupy a worthy place in the diet. In addition, they are prescribed (mainly in the winter-spring period, when one’s own reserves are depleted) in the form of medicinal forms.
The patient will also have to forget about drugs that disrupt platelet function and increase bleeding (NSAIDs, chlorpromazine, fibrinolytics, anticoagulants, penicillin antibiotics, etc.). The direct anticoagulant heparin is used exclusively for health reasons (in case of development of disseminated intravascular coagulation syndrome).
For the treatment of hereditary types of thrombocytopathies accompanied by significant bleeding in children and adults (essential athrombia, disaggregation thrombocytopathy, mild and moderate von Willebrand disease), aminocaproic acid in a dose calculated according to weight. The positive effect of the drug is especially pronounced for bleeding from the uterus and nasal passages.
Aminocaproic acid is also useful in the case of acquired pathology of primary hemostasis (thrombocytopathies that occur after massive blood transfusions, medication, uterine bleeding caused by a decrease in estrogen).
Aminocaproic acid is prescribed intravenously, but it should be borne in mind that its introduction into a vein can provoke thrombosis or disseminated intravascular coagulation, so the drug is used carefully and only in emergency cases when bleeding threatens the patient’s life.
To treat disorders of platelet-vascular hemostasis of various origins, in addition to aminocaproic acid, its “relatives” are often used - tranexamic acid , which has tablet forms (cyclocapron) and para-aminomethylbenzoic acid (PAMBA).
They remind platelets of their immediate tasks (to stick together and form conglomerates) and contraceptives ( synthetic hormonal contraceptives ), they help to significantly reduce bleeding in symptomatic (acquired) forms. However, as they say, there is another side to the coin. The positive hemostatic effect of oral contraceptives is counteracted by the risk of thrombosis and provocation of DIC syndrome. The simultaneous use of contraceptives and aminocaproic acid may also be life-threatening. Here, it is unlikely that it will be possible to do without the prevention of ICE.
Other drugs prescribed for various types of thrombocytopathies:
- ATP (adenosine triphosphate) together with magnesium sulfate (MgSO4). Such therapy is effective in cases of partial disaggregation thrombocytopathy, which occurs with a violation of the “release reaction”, and is practically useless in the case of essential athrombia, Glyantsman thrombasthenia, Herzmansky-Pudlak syndrome and “gray platelet” syndrome;
- Adroxon - increases the functional abilities of blood platelets, helps normalize microcirculatory hemostasis, the drug is prescribed for any type of pathology, including those combined with DIC syndrome;
- Dicynon (intramuscular, subcutaneous, orally);
- Bradykinin antagonists (anginin, parmidin);
- Synthetic derivatives of vasopressin (remestil, terlipressin), which cause an increase in the activity of von Willebrand factor and are combined with other hemostatic agents (aminocaproic acid, cryoprecipitate);
- Antihemophilic plasma and cryoprecipitate (used for von Willebrand disease, as the main replacement treatment, as well as for Glyantsman thrombasthenia, essential atrombia, acquired thrombocytopathies).
To stop minor bleeding in accessible areas, local treatment can be used, for example, in the following sequence: cold 5% solution of aminocaproic acid → hemostatic gel containing fibrinogen, thrombin and calcium chloride, → adroxon → collagen. Herbal infusions that have a hemostatic effect may be useful.
It should be noted that blood transfusions in most cases do not provide a hemostatic effect and do not bring any benefit, and transfusions of large volumes of blood also complicate the situation, since they lead to the development of disseminated intravascular coagulation with thrombocytopenia of consumption. True, with massive blood loss it is still difficult to avoid transfusions of fresh frozen plasma, platelet concentrate, and red blood cells.
Reasons for the development of the disease
The causes of thrombocytopenia in adults lie in gene mutations, because hereditary pathology appears in this case. Underlying diseases and other factors can lead to the development of secondary (symptomatic) thrombocytopathy. These are, for example, significant transfusions of platelets or other blood cells. But the most common factor leading to impaired aggregation of coagulation factors is impaired absorption or deficiency of vitamin B12.
In addition, such a disease often occurs against the background of hemoblastosis (for example, disaggregation thromocytopenia). Often the pathology develops with uremia. This is a state of constant autointoxication resulting from chronic severe renal failure) Other forms of secondary thrombocytopathies appear in the following diseases:
- DIC – syndrome;
- cirrhosis, tumors and parasitic liver lesions;
- scurvy;
- endocrine disorders, in particular hyperproduction of estrogen and hypothyroidism;
- radiation sickness.
Drug-induced and toxigenic secondary thrombocytopenias are classified into a separate group. They occur as a complication after a long course of taking aspirin, some antibiotics (for example, penicillins and nitrofurans), and cytostatics. Often, platelet aggregation disorders develop against the background of multiple myeloma and myeloproliferative disorders. In some cases, it is not possible to establish the exact cause of the pathology, then the diagnosis of “unspecified thrombocytopathy” is recorded in the patient’s card.
There are reasons that sharply increase the risk of developing and increasing blood circulation. Particular attention should be paid to patients who may exhibit hereditary thrombocytopathy. Self-monitoring of your own condition will allow you to neutralize the disease at the very beginning and avoid unpleasant complications. Bleeding is caused by:
- foods containing large amounts of vinegar;
- all alcohol-containing drinks;
- drugs that have an anticoagulant effect. Some anti-inflammatory medications;
- most physical procedures, especially ultra-high frequencies, ultraviolet radiation;
- prolonged exposure to the scorching sun.
Determination of genetic predisposition to this type of pathology should be carried out in the early stages of pregnancy. If there are cases of thrombocytopenia in the family, the woman should consult a geneticist and, if necessary, undergo certain tests. After birth, the child must be under medical supervision. Subsequently, it is necessary to register with a hematologist, which will avoid complications and irreversible consequences of the disease.
Causes
Main causes of the disease:
- genetic disorders of the structure of platelet walls;
- malignant blood lesions;
- severe dysfunction of the liver and kidneys;
- vitamin B deficiency;
- radiation sickness;
- scurvy;
- DIC syndrome;
- blood transfusion.
The disease can manifest itself during long-term therapy with NSAIDs, analgesics, antibiotics and tranquilizers.
Diagnostics
Diagnosis, especially if it is thrombocytopathy in children, begins with a detailed analysis of the medical history and life of the child or adult. A physical examination will help notice pale skin, the presence of large bruises, and they can be of different colors: from bluish-black to pale yellowish. The pulse rate in such patients is increased, but blood pressure (BP) levels are reduced. Next, laboratory diagnostics are prescribed:
- Complete blood count, in which the platelet count will be normal or only slightly reduced. But the levels of hemoglobin and red blood cells will “fall” below the minimum norm.
- Clinical urine analysis reveals an increased content of red blood cells. The doctor may suspect renal pathology.
- Blood biochemistry must necessarily include a test for cholesterol (fat content), glucose, creatinine, electrolyte composition, and uric acid. These indicators will indicate the original causes, complicated by thrombocytopathy.
- If a diagnostic bone marrow puncture was performed, the puncture is examined in detail cytologically.
- Trephine biopsy: a special type of pathology detection that requires the intervention of several specialists.
Naturally, the examination begins with the simplest clinical tests, and then moves on to more detailed diagnostic tests. Separately, you need to dwell on the hemostasiogram - a unique laboratory technique that allows you to visualize and evaluate all stages of blood coagulation. With this analysis, types of disturbances in aggregation, retraction, adhesion, and shifts in the time of blood clotting are clearly visible. Additionally, diagnostics includes a number of mandatory tests:
- using a rubber band, which is tied on the forearm for 5 minutes. After removing the device, patients with thrombocytopathy will have a clear bluish stripe;
- reaction to a pinch;
- finger pricking with further assessment of the duration of bleeding;
- We place the cuff from any type of tonometer on the forearm of either hand. Next, we pump air to 100 mm. rt. st, time for 5 minutes, remove the cuff and evaluate the color of the skin under it. If pinpoint hemorrhages appear, then thrombocytopathy can be suspected with high probability.
When the basic diagnostics are completed and all the results of instrumental studies are evaluated, it is possible to invite specialists for a consultation. Usually an examination is required by a urologist, therapist, pediatrician (if it concerns children), gynecologist (if the patient is pregnant), etc. Collegiality when making a diagnosis will help avoid mistakes and give the patient confidence that the diagnosis was correct. To determine the exact form of thrombocytopathy, specific laboratory tests are required. For example, Bernard-Soulier syndrome is characterized by thrombocytopenia with a predominance of macrocells and the absence of von Willebrand factor - dependent aggregation with ristocetin with normal activity and concentration of von Willebrand factor itself.
THROMBOCYTOPATHY
THROMBOCYTOPATHY
(platelet[s] + Greek pathos suffering, disease) - a group of hemorrhagic diathesis caused by the qualitative (functional) inferiority of platelets and characterized by a violation of vascular-platelet hemostasis - a multi-stage reaction leading to the formation of a hemostatic plug. With T., the platelet count (see) is usually normal.
T. are a heterogeneous group of diseases due to pathogenetic mechanisms. They are distinguished by rather uniform and meager symptoms; forms predominate, manifested by hemorrhages of predominantly microcirculatory type - petechiae (see) and ecchymosis (see Hemorrhage), less often - subcutaneous hemorrhages, as well as gingival, nasal and uterine bleeding, menorrhagia, etc. T. are distinguished hereditary and congenital, as well as acquired , or secondary, which are accompanied by other patol. processes (hemoblastosis, liver disease, kidney disease, etc.). According to V. A. Germanov (1966), thrombocytopathies occupy first place in frequency (36%) among hereditary hemorrhagic diathesis (see).
Austin (J. N. Austin), Pepper (O. N. P. Pepper) in 1913 and Hess (AF Hess) in 1916 observed purpura characteristic of Werlhof's disease (see Thrombocytopenic purpura) with a normal platelet count. In 1918, E. Glanzmann described hereditary hemorrhagic thrombasthenia, in which a violation of blood clot retraction (see Retraction) was explained by a deficiency in platelets of a hypothetical substance - the so-called. retractozyme. In 1926, E. A. Willebrand described angiohemophilia (see), in which platelet dysfunction is also noted, leading to impaired thrombus formation and prolongation of bleeding time (see Bleeding time).
In 1948, J. Bernard and JP Soulier proved the existence of a new syndrome, which they called thrombocytodystrophy and characterized by a violation of the use of thrombin (see) and the presence of giant platelets (see). In 1965, Ulutin (O. N. Ulutin) and in 1967, Weiss (H. J. Weiss) showed the presence of impaired platelet release reactions in T. (see). Ulutin called this form primary (functional) thrombocytopathy.
In 1968, E. J. Bowie and S. A. Owen, and in 1970, Holmsen and Weiss identified among T. deficient thrombocytopathies, or so-called. diseases of insufficient accumulation, which are characterized by a decrease in the content of ATP, ADP, serotonin, adrenaline, platelet factor 3 and other active substances in platelets.
T. are divided into isolated, characterized by one functional defect, for example, a violation of platelet aggregation, and combined, in which several functional disorders of platelets are noted, for example, adhesion is simultaneously impaired (see Adhesion, vol. 20, additional materials) and aggregation platelets.
There are a number of classifications of T. The most complete is the classification of 3. S. Barkagan (1973), based on the morphofunctional characteristics of platelets. Below are the most common forms of T. in accordance with this classification.
Thrombocytopathies with predominantly impaired aggregation function
. Among these forms, the most common are thrombasthenia Glanzmania and essential athrombia. Glanzmann's thrombasthenia is inherited in an autosomal recessive manner. The disease usually appears at a young age. Bleeding (hemorrhages in the skin, mucous membranes, etc.) is usually moderate. The slightest damage to the skin and mucous membranes leads to prolonged bleeding, nasal and uterine bleeding, and a tendency to hypermenorrhea are also noted. Hemorrhagic syndrome is more pronounced at a young age and in women. Of particular danger are bleeding after tonsillectomy, as well as cerebral hemorrhages. The number of platelets and their sizes are normal, morphol. They show no abnormalities. Bleeding time is significantly prolonged, and blood clot retraction is slowed. Platelet aggregation under the influence of ADP, adrenaline, collagen, serotonin and prostaglandins is not observed. Platelets normally adhere to the subendothelium of vessels, but do not aggregate with neighboring platelets, as a result of which an effective hemostatic plug does not form at the site of damage to the vessel wall. The cause of the aggregation failure is unknown. In some patients, the fibrinogen content in platelets is reduced (Glanzmann's thrombasthenia type I), in others the fibrinogen content is normal (Glanzmann's thrombasthenia type II); There is also a decrease in the antigen associated with coagulation factor VIII. A deficiency of specific glycoproteins was detected in platelet membranes.
Essential athrombia (disaggregative thrombocytopathy without impaired release reaction, according to Barkagan) in terms of wedge, manifestations and nature of platelet dysfunction does not differ from Glanzmann’s thrombasthenia. With this T., the sensitivity of platelets to ADP is slightly reduced, and blood clot retraction is normal.
Forms with a violation of the release reaction and the second phase of platelet aggregation
are inherited in an autosomal recessive manner and, less commonly, in an autosomal dominant manner. Wedge, the picture of these T. is the same. Patients experience slight bleeding, manifested by hypermenorrhea and nosebleeds. The skin and mucous membranes are easily injured. Platelets do not aggregate with collagen. In patients with this form of T., the reaction of platelet release is impaired (with aggregation caused by collagen, ADP, adrenaline). This is combined with other congenital defects observed, for example, in Wiskott-Aldrich syndrome (see Wiskott-Aldrich syndrome), Chediak-Higasn syndrome (hereditary neutropenia with hyopigmentation, frequent inf. complications, hepatosplenomegaly and other phenomena), Herzhmansky- Pudlak (see Herzmansky - Pudlak syndrome). Herzmansky-Pudlak syndrome is usually inherited in an autosomal recessive manner. With it, the degree of bleeding is small, the bleeding time is somewhat prolonged. The skin and mucous membranes are easily vulnerable, ecchymosis, nosebleeds and menorrhagia are observed. The tendency to increased bleeding persists throughout life. This pathology is combined with albinism (primarily of the skin and eyes) and the presence of pigmented macrophages in the red bone marrow.
Forms with a violation of the release reaction and the second phase of aggregation include T. associated with insufficiency of prostaglandin synthesis (see). They are observed in some people taking acetylsalicylic acid. At the same time, in the wedge, the picture is dominated by various types of purpura, menorrhagia, and less often - gastric bleeding. If platelets contain normal levels of ADP, ATP and serotonin, stimulation with collagen, ADP or adrenaline does not cause a release reaction. It is believed that acetylsalicylic acid inhibits the specific enzyme cyclooxygenase, which is necessary for the conversion of arachidonic acid into intermediate products of prostaglandins and thromboxanes, which, in turn, serve as intermediaries for the release of adenonucleotides and serotonin. The platelet release reaction triggered by collagen or thrombin depends on the synthesis of cycloendoperoxides (prostaglandins and thromboxane A2), which are strong aggregating agents. However, the mechanism of action of acetylsalicylic acid on platelets is complex and insufficiently studied. There is evidence of its damaging effect on the ultrastructure of platelets and the inhibition of a number of their functions.
Thrombocytopathies with partial disorders of certain types of aggregation without pathology of the platelet release reaction
characterized by platelet dysfunction - impaired platelet aggregation under the influence of collagen (collagen-aggregation), large and small doses of ADP (ADP-aggregation), adrenaline, small doses of thrombin (thrombin-aggregation), ristocetin (ristocetin-aggregation). Among these forms of T., the most studied is the May-Hegglin anomaly, or the May-Hegglin syndrome (see Thrombocytopenia), in which ristocetin aggregation is impaired.
Insufficient accumulation diseases are characterized by a significant decrease in the content of ADP, to a lesser extent ATP, and a decrease in the content of serotonin in platelets, as well as an abnormality in the ATP ADP ratio. Impaired accumulation of these substances is observed in Wiskott-Aldrich, Chediak-Higashi and Herzmansky-Pudlak syndromes. With Wiskott-Aldrich and Chediak-Higashi syndromes, thrombocytopenia is also observed (so-called thrombocytopenic thrombocytopathy); however, the degree of bleeding is inadequate to reduce the platelet count. Both syndromes with this pathology are often severe (severe bleeding is noted, for example, from the gums, nosebleeds, etc.) and have an unfavorable prognosis due to the addition of infection. In Herzmansky-Pudlak syndrome, the platelet count is normal; the syndrome can be isolated and not combined with any of the above syndromes; the disease is caused by a deficiency of type I dense bodies in platelets, which leads to impaired accumulation of serotonin and adeine nucleotides.
In case of a storage disease - type I glycogenosis (see Glycogenosis), as well as in case of deficiency of the enzyme fructose-1,6-biphosphatase, a disturbance in the synthesis of nucleotides is observed. At the same time hron. hypoglycemia is combined with minor bleeding, nosebleeds and slight vulnerability of the skin and mucous membranes due to impaired nucleotide release and platelet aggregation.
Forms with deficiency and decreased availability of platelet factor 3
represent T., which are characterized by insufficient coagulant activity of platelets. This leads to disruption of the interaction of platelets with plasma coagulation factors, i.e., disruption of the blood coagulation process. Such T. occurs with nasal and gingival bleeding, ecchymosis, menorrhagia, bleeding after tooth extraction, etc. Lab. research methods reveal an increase in bleeding time, a decrease in prothrombin consumption (see), an increase in the kaolin clotting time of blood plasma, etc.
Thrombocytopathies with a predominant violation of platelet adhesiveness.
The causes of such disorders can be hereditary connective tissue diseases, for example. Ehlers-Danlos syndrome (see Desmogenesis imperfecta), deficiency of plasma proteins necessary for platelet adhesion, for example. von Willebrand disease (see Angiohemophilia), platelet membrane abnormality (eg, Bernard-Soulier syndrome). With Ehlers-Danlos syndrome, mild signs of bleeding are observed due to collagen abnormalities (no adhesion of normal platelets occurs). In addition, platelet deficiency is revealed due to a violation of the release reaction.
Bernard-Soulier syndrome (macrothrombocytodystrophy) is inherited in an autosomal recessive manner. The disease is characterized by prolonged bleeding time, the presence of giant platelets in the blood with their normal or slightly reduced number, and platelet factor 3 deficiency. The severity of hemorrhagic symptoms depends on the prolongation of bleeding time and the number of giant platelets. Platelet aggregation in patients under the influence of ADP, collagen and thrombin is not impaired; platelets do not aggregate with ristocetin or with bovine fibrinogen preparations containing coagulation factor VIII. In platelet membranes, the content of a specific glycoprotein that normally reacts with blood coagulation factor VIII and ristocetin and is (presumably) a receptor for them is reduced. An abnormality of the platelet membrane can lead to shortened platelet lifespan and thrombocytopenia, as well as impaired binding of plasma clotting factors. One manifestation of this is decreased prothrombin consumption, which in turn reflects a decrease in available platelet factor 3.
Differential diagnosis of Bernard-Soulier syndrome is carried out with von Willebrand disease, in which there are no giant platelets, and with the May-Hegglin anomaly, also characterized by giant platelets, but in combination with Dele's bodies in neutrophils.
Acquired, or secondary, thrombocytopathies are caused by secondary dysfunction of platelets in myeloproliferative diseases (see), hron. renal failure, accompanied by uremia, disseminated intravascular coagulation (see Hemorrhagic diathesis), etc. In a number of conditions, platelet dysfunction occurs when platelets interact with macromolecules (paraproteins, dextran) and certain drugs (anti-inflammatory drugs, penicillin, phenothiazide , alpha-blocker, prostaglandin E and others). Acquired T. can also occur with massive blood transfusions. In a number of diseases, the mechanism of platelet dysfunction is complex (for example, DNA synthesis, platelet maturation, and the activity of their enzymes are impaired). With acquired T. platelet anomalies are not clearly expressed.
The diagnosis of thrombocytopathies is established on the basis of a wedge, picture, family history, hemogram (see), myelogram (see), the state of vascular platelet hemostasis (see), as well as morphology, function, population composition and biochemical. platelet characteristics (see). Differential diagnosis must be carried out, first of all, with thrombocytopenia (see) and hemophilia (see). It should be borne in mind that hereditary T. is characterized by a triad of symptoms: the hereditary nature of the disease (often in combination with other genetic anomalies, for example, albinism); stable functional, morphological and biochemical changes in platelets that are not eliminated by normalizing their number in the blood; discrepancy between the severity of bleeding and the degree of existing thrombocytopenia.
Treatment of all forms of thrombocytopathies is complex (taking into account the etiology, pathogenesis and form of the disease or syndrome) and is mainly symptomatic. Treatment of acquired T. consists of eliminating the underlying disease or provoking factor. As a preventive measure, it is prohibited to drink alcohol and consume vinegar with food, as well as home-canned food prepared with the use of chemical salicylates. It is recommended to take large doses of vitamins C, P and A, and include peanuts in the diet, which improves microcirculatory hemostasis and platelet function. When treating intercurrent diseases in patients with hereditary forms of T., drugs that impair platelet function or affect blood clotting (salicylates, brufen, indomethacin, butadione carbenicillin, angina, clofibrate, persantine, aminazine, indirect anticoagulants, fibrinolytics) should not be prescribed. For disaggregation T., von Willebrand's disease, as well as for acquired T. (for example, after transfusion and drug therapy, hypoestrogenic uterine bleeding), aminocapronic acid is indicated.
Adrenochrome monosemicarbazone (adroxon) has a good hemostatic effect for any type of tumor; dicinone is less effective. Transfusions of platelet mass (see) are indicated in emergency cases (profuse bleeding, preparation for surgery) or when hematopoiesis is suppressed, in particular the megakaryocyte lineage.
Bibliography:
Barkagan 3. S. Hemorrhagic diseases and syndromes, p. 59, M., 1980; Hemorrhagic platelet diseases, ed. V. A. Germanova, Kuibyshev, - 1978; Germanov V. A. and Piksanov O. N. Erythrocytes (erythron), platelets (platelet-megakaryocyte system), leukocytes (leukemia), Kuibyshev, 1966; Odesskaya T. A. On the issue of bleeding of unknown etiology. (Thrombocytopathies of congenital and acquired nature), Probl. hematol. and overflow, blood, vol. 19, no. 5, p. 27, 1974; Problems and hypotheses in the doctrine of blood coagulation, ed. O. K. Gavrilova, M., 1981; Guide to Hematology, ed. A. I. Vorobyova and Yu. I. Do-rie, p. 104, 483, M., 1979; Smolyanitsky A. Ya. and Bokarev I. N. Methods for diagnosing thrombocytopathies, Laboratory. case, No. 4, p. 195, 1974, bibliogr.; Shabalov N.P. Hereditary thrombocytopathies, Probl. hematol. and overflow. blood, vol. 21, no. 7, p. 40, 1976, bibliogr.; Shitikova A. S. Classification of functional disorders of platelets, ibid., vol. 26, no. 3, p. 49, 1981; Yurlov V.M. Some issues of classification and differential diagnosis of hemorrhagic thrombocytopathies, ibid., vol. 23, no. 6, p. 37, 1978, bibliogr.; Platelets, recent advances in basic research and clinical aspects, ed. by ON Ulutin, NY, 1975; W eiss HJ Platelet physiology and abnormalities of platelet function, New Engl. J. Med., v. 293, p. 531, 1975.
Yu. N. Tokarev.
Symptoms of the pathological condition
Symptoms of a pathological condition may not attract attention for a long time. Due to the wavy or erased course, the clinical picture of the disease can last for several weeks, or even years. The congenital form of the disease can be noticed in the maternity hospital, after blood tests are performed on the newborn. All symptoms can be conditionally combined into the following syndromes: hemorrhagic and anemic.
Manifestations of hemorrhagic syndrome
Symptoms of hemorrhagic syndrome with thrombocytopathy are manifested by excessive bleeding: heavy uterine bleeding during menstruation begins to bother you, gums bleed, minimal trauma causes profuse bleeding. Sometimes just scratching your nose or gently brushing your teeth is enough to cause heavy bleeding. In severe cases, hematuria (blood in the urine), occult blood in the stool and bloody vomiting may develop. Sometimes the tendency to form subcutaneous hematomas and bruises attracts attention.
Manifestations of anemic syndrome
Anemia is a pathological condition of the body in which tissues experience chronic oxygen starvation caused by a decrease in hemoglobin. Hemoglobin is a “carrier” that delivers oxygen to tissue cells. If this process is disrupted, then redox reactions are distorted and almost all body systems malfunction. All anemic syndromes have almost the same symptoms:
- pale skin;
- poor general health: low performance and resistance to stress, absent-mindedness, dizziness, frequent fainting or near-fainting states;
- after minor physical activity, shortness of breath, increased heart rate, and aching diffuse pain in the heart area attract attention.
Causes of thrombocytopathy
The causes of congenital thrombocytopathies come down to disturbances in the gene structure. These anomalies lead to a malfunction in the body at the level of platelet hemostasis. Inheritance of such anomalies can occur either in an autosomal recessive or an autosomal dominant manner.
The causes of acquired thrombocytopathies are as follows:
Mixed type hemoblastoses, disseminated intravascular coagulation syndrome and disaggregation hyporegenerative diseases of the hemostatic system can cause the development of thrombocytopathy.
The triggering factor leading to platelet failure can be myeloproliferative diseases and essential thrombocythemia.
Drug therapy for the disease
Treatment of thrombocytopathy begins only after evaluation of these diagnostic procedures. Treatment with medications is a responsible and main stage of therapy. The treatment regimen includes:
- fibrinolysis inhibitors: Pamba, Exacil, Ingitril.
- Drugs that increase the rate of blood clotting. Take for a short time, only during periods of heavy bleeding.
- Contraception in women. As a result of their positive effect on platelets, medications of this pharmaceutical group stabilize uterine bleeding during menstruation.
- Drugs that reduce the lumen of the bloodstream. They are the medications of choice for quickly stopping bleeding.
- Metabolites. Qualitatively improve metabolic reactions inside platelets, increasing the quality of blood cells. For example, folic acid significantly improves hematopoiesis.
- Vitamins of group B (Milgamma compositum, Angiovit), group A (Retinol), group C (ascorbic acid), group PP (Nicotinic acid, Nicotinamide, Niacin). Vitamin preparations such as Ascorutin, Centrium, Teravit, Vitrum.
If drug therapy is ineffective, additional examination is prescribed. The issue of a person’s ability to work is resolved and treatment tactics are developed. If all methods do not produce stable remission, then scalpel intervention is indicated.
Development mechanism
The development of the disorder is based on a group of possible moments. In more detail:
Hereditary predisposition, defects of certain genes
According to statistics, the deviation in question occurs in 5-10% of cases of the total disorder of the hematopoietic and hemostasis system.
According to various estimates, genetically determined thrombocytopathies account for up to 70% of clinical situations.
In this case, mutations are noted that exclude the possibility of cure. All that remains is to correct the symptoms.
Hereditary forms are accompanied by rapid progression of the condition, critical disruptions in the functioning of all organs and systems.
Decreased production of coagulation factors, platelet adhesion and aggregation
As a result of hypovitaminosis, for example, deficiency of B12 or B9 (megaloblastic anemia), other disorders such as DIC, ongoing autoimmune inflammatory processes (as an option - systemic vasculitis).
In both cases, the disorder develops rapidly. However, hereditary forms of thrombocytopathy provoke the main symptoms immediately, from childhood.
Due to the lack of specific signs, even experienced doctors cannot always immediately determine the essence of the pathological process. False diagnoses are made and ineffective therapy is provided. This could cost the patient's life.
Other methods of traditional treatment
Modern types of operations are a chance to lengthen a person’s life and improve their quality of life. Scalpel interventions are often performed only in a hospital setting, subject to the basic rules of preoperative preparation and management of the postoperative period. Here are the most common types of operations:
- Splenectomy;
- Suturing a bleeding vessel. Sometimes a blood line prosthesis is required.
- Puncture of the articular cavity. Manipulation becomes necessary when there is hemorrhage in the joint capsule. If there is no effect and the joint block increases, then it is replaced with an artificial analogue.
Only an integrated approach can cope with the complications of thrombocytopathies. If joint contracture increases, and to improve the functioning of the whole body, feasible physical therapy is prescribed. The frequency, amplitude and time of performing the exercises are determined by the instructor. You can do a complex of exercise therapy at home, which is convenient during periods of postoperative rehabilitation.
How is it diagnosed?
If there is bleeding, the patient first turns to a therapist, who refers to specialists of other profiles. A consultation with a hematologist and a doctor with a more specialized profile is required: gynecologist, ENT specialist, dentist, etc., depending on the location of the bleeding.
To make a correct diagnosis, the patient must undergo examinations such as:
- endothelial test;
- blood tests for platelet content and function (aggregation, adhesion, blood clot retraction), as well as general;
- determining the duration of bleeding and coagulation;
- assessment of hematopoietic function (if necessary);
- rarely - trephine biopsy.
Principles of emergency care
Every parent raising a child who has congenital (hereditary) thrombocytopathy should know how to stop bleeding. There must be medicines and available supplies in the house (bandage, cotton wool, medical tourniquet, etc.). Here are the principles of emergency care for nosebleeds:
- Calm an adult or child.
- Tilt the victim's head back.
- Place an ice pack, cold compress, a bottle of water from the refrigerator, ice cream, etc. on the bridge of your nose.
- Place a piece of hemostatic (hemostatic) sponge into the nasal cavity.
- Additionally, gauze swabs are inserted into the nostrils, but care is taken that the patient does not push them into the nasopharynx.
If bleeding occurs in the joint cavity, then try to immediately immobilize the bone joint. To do this, apply a pressure bandage, and the limb is immobilized. Ice is applied to the area of the affected joint. Further treatment is continued after consultation with a traumatologist. Hemorrhage in the orbit is a reason for an urgent visit to the ophthalmology department. An ophthalmologist treats patients with eye pathologies.
What treatment is prescribed
If the diagnosis is confirmed and a specific type of thrombocytopathy is determined, then treatment and clinical recommendations are selected for the patient.
Depending on the type of disease, 1 of 3 treatment options are used:
- etiotropic, which is aimed at eliminating the cause and is used only for acquired thrombocytopathies;
- preventive, supporting the patient’s condition, preventing the pathology from developing further;
- urgent, aimed at quickly eliminating the problem, it may even involve surgery.
In addition, the patient is prescribed a special diet. Nutrition can play a significant role in the treatment of thrombocytopathy . So, alcohol and vinegar in any form are completely excluded. It is recommended to increase the content of animal protein in the diet, enrich food with foods containing vitamins C, P, A. Peanuts are healthy.
As for medications, a course of vitamin and mineral complex is prescribed. Drugs that stimulate metabolism and vasoconstrictors are added, for example, fibrin film and aminocaproic acid are prescribed.
Folk remedies can also be useful. Teas, herbal infusions and infusions that improve blood clotting and its composition are recommended for consumption.
Traditional therapy and diet therapy
Thrombocytopathy has long been fought with herbal remedies. For example, if your gums are bleeding excessively, you should brew a strong decoction of oak and linden blossom. After the broth has cooled to a warm state, carefully rinse the bleeding gums several times a day. Herbs can be processed year-round using dried or pharmaceutical raw materials. Here are a few more recipes from the people.
Rec. No. 1
If the bleeding is in the organs of the digestive system or in the area of the uterus, then take a pharmaceutical tincture of water pepper. 1 tbsp. A spoonful of the drug is mixed with two glasses of water. You should drink it throughout the day, after dividing it into equal portions. Take after a heavy meal. The help water pepper provides is quite noticeable, which is why this inconspicuous plant is loved.
Rec. No. 2
If there is thrombocytopenia, then take 8 tablespoons of crushed viburnum bark, boil for 30 minutes in two glasses of water, carefully closing the mug. After this, add enough water to return the drug to its original volume. One glass is enough for 1 day of use.
Diet therapy
The nutrition of a patient with thrombocytopathy plays a huge role. Types of diet are selected so that it contains a maximum of vitamins, the entire range of microelements, and a minimum of heavy fats. Peanuts and peanut butter, almonds, walnuts, and young leaves of red grape varieties stabilize platelet quality well. Lingonberries, plantain, green tea are excellent substitutes for regular black tea. Triple attention to diet is necessary if pregnancy occurs.
Disaggregation thrombocytopathy
Disaggregation thrombocytopathy is in the second group of hemorrhagic diathesis, manifested by poor blood clotting. Heredity and other reasons lead to this.
The hereditary form of the disease is divided into 2 groups:
- Glanzmann's thrombasthenia.
- May-Hegglin anomaly.
The first form occurs without a “release process” and is transmitted with both dominant and recessive genes. Clinically manifests itself in the form of small point hematomas.
This form occurs more often in girls who experience long periods of bleeding from the uterus, the function of which is unknown. Subsequently, signs of posthemorrhagic diathesis appear, which manifests itself in severe form.
A characteristic clinical manifestation of this disease is hemorrhages in the retina or brain.
The May-Hegglin anomaly occurs without disruption of the “release process,” but it is characterized by external and internal changes in platelets.
No obvious clinical manifestations are observed; in laboratory studies, this form of the disease can be detected in the form of thrombocytopenia and huge metric parameters of the cells themselves.
A few words about complications and prevention
If the root cause of thrombocytopathies cannot be eliminated and blood pathology increases, then severe complications may occur: iron deficiency anemia, sudden changes in the immune (barrier) system, paralysis, blindness, and the development of anemic coma. Patients are concerned about the general deterioration of health, the functioning of internal organs is disrupted, the liver and kidneys are especially vulnerable. Almost a third of patients complain of frequent and heavy bleeding from the nasal vessels.
Primary prevention begins with a consultation at a medical genetic center, which couples who have a congenital disease must undergo. Be sure to exclude self-medication and uncontrolled use of medications of any pharmacological group. Bad habits, occupational hazards and risks of radiation exposure are taken under strict control. In no case should you neglect preventive examinations, routine tests and other instrumental preventive studies, and eradicate other causes of the appearance in patients with thrombocytopathy.
Symptoms
The clinical picture is determined by the development of typical disorders. They correspond to disorders of normal coagulation.
- Frequent nosebleeds. Regardless of the influence of third-party factors. At the same time, blood pressure remains normal or lower than that, and there are also no mechanical influences or injuries.
The intensity of the process is so great that in some cases it is impossible to do without medical help right away. Tamponade and the use of drugs to restore normal condition will be required.
Read about other causes of nosebleeds (frequent and episodic) in adults here, and in children here.
- Bruises and hematomas on the body are a mandatory, but not characteristic sign of thrombocytopathy. Arranged in random order. This is the result of a violation of the anatomical integrity of blood vessels and small capillaries (hemorrhagic syndrome). The dimensions can be significant: up to several centimeters and even more.
As the manifestation progresses, it becomes more and more obvious, accompanied by a gradual increase in the number of bruises. In this case, there is no pain at the site of hematoma formation, because there was no mechanical factor - tissue damage.
- Weakness. Impairment of normal working capacity. A person tends to lie down, there is no strength to do everyday work. Muscle asthenia is present. The muscles become sluggish, it is impossible to move even short distances.
- Drowsiness. During the daytime, at night. Constantly. Regardless of the amount of rest. This is a typical manifestation of iron deficiency anemia, which develops in almost all patients with thrombocytopathy, even in the early stages. The secondary pathological process progresses rapidly, parallel to the formation of the main disorder.
- Tachycardia. Increased heart rate. How pronounced it is depends on the degree of anemia and hemoglobin deficiency.
Cardiac structures do not receive enough oxygen and nutrients, and cannot work at the same rhythm. Intensification occurs as an adaptive mechanism. But this does not produce results.
- Falling blood pressure levels. 10-30 mmHg less than the individual functional norm for a particular person.
- Dyspnea. Including without any physical activity. Accompanied by the inability to work, move, or climb stairs. The person is in a state of forced rest.
- Sweating. Hyperhidrosis. Also a result of anemia.
- Frequent fainting. Insufficient nutrition and oxygen supply also affects the brain. Syncopal episodes become more frequent as the underlying diagnosis progresses.
- Menorrhagia. In girls and adult women. Heavy menstrual bleeding, as well as the outpouring of liquid connective tissue, is possible regardless of the cycle.
Attention:
This is a deadly symptom that cannot but affect the general condition, aggravates the course of anemia, and provokes deadly complications in patients. Immediate hospitalization to a gynecological hospital is required.
- Massive bleeding. Gastrointestinal, pulmonary, others. They pose a critical threat to the patient's life. They begin spontaneously, without visible provocateurs.
- Impossibility of normal surgical interventions due to the general fragility of blood vessels. Inability to achieve adequate spontaneous hemostasis. This is a big problem because even dental treatment becomes an overwhelming task.
- Objectively, occult blood is detected in stool tests, what can we say about constant minor bleeding. Hematuria is traces of red blood cells in the urine, which should not be the case.