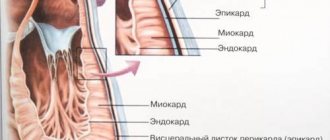
Cardiac amyloidosis (or amyloid cardiopathy) is a pathological condition accompanied by the deposition in the intercellular space of cardiac tissue of amyloid - an insoluble substance with a b-fibrillar structure, consisting of proteins associated with polysaccharides and formed during a metabolic disorder. The accumulation of such a protein-saccharide complex in the cardiac muscle, pericardium, endocardium, aortic walls and coronary vessels leads to myocardial hypertrophy, disruption of the normal contractility of this vital organ and hemodynamics, valve defects, hypotension and arrhythmias.
Cardiac amyloid damage can be observed in different types of protein metabolism disorders - light chain amyloidosis, systemic cyanide, familial or secondary amyloidosis. In the generalized course of amyloidosis, the heart becomes one of the first targets. In addition to it, other organs may subsequently be affected - the liver, spleen, kidneys, lungs, intestines, etc. However, in some cases, isolated (local) atrial amyloidosis may be observed.
So far, scientists have not been able to learn how to completely cure this rare disease. However, they have developed drug and surgical treatment regimens that help prolong the life of patients with this diagnosis.
In this article we will provide you with information about the causes, manifestations, mechanism of development, prognosis, methods of diagnosis and treatment of cardiac amyloidosis. This knowledge will help you suspect the development of this dangerous disease in time, and you will make the right decision about the need to see a doctor.
Causes
The primary cause of the development of cardiac amyloidosis is a disorder of protein metabolism, accompanied by the formation of amyloid. Such a metabolic failure can be provoked by hereditary causes, leading to the development of familial amyloidosis. This disease is more often observed among the population of Mediterranean countries and is transmitted in an autosomal dominant manner.
Another type of cardiac amyloidosis is secondary amyloidosis, which can occur with a long-term course of the following ailments and conditions:
- syphilis;
- tuberculosis;
- Crohn's disease;
- rheumatoid arthritis;
- ankylosing spondylitis;
- psoriatic arthritis;
- actinomycosis;
- bronchitis;
- bronchiectasis;
- lymphogranulomatosis.
Sometimes the cause of cardiac amyloidosis is long-term renal failure. In such cases, hemodialysis may become a trigger for protein metabolism disorders.
In more rare cases, cardiac amyloidosis is detected in older people. It is assumed that amyloid deposition is provoked by many malfunctions in the functioning of the aging body, caused by various pathologies “accumulated over the years” - disturbances in the functioning of the central nervous system, pancreas, liver, etc. As a rule, in such cases, cardiac amyloidosis is detected only after an autopsy.
Development mechanism
Amyloid deposition in cardiac tissue is observed in almost 100% of patients suffering from familial, idiopathic or advanced cyanide amyloidosis. Mostly this protein-saccharide substance is deposited in the myocardium and less often accumulates in other layers of the heart.
In the heart muscle, it occupies the space between myofibrils (fibers of muscle tissue) and compresses the vessels of the heart. Because of this, the myocardium becomes compacted and inelastic. The cardiac chambers change little.
In the senile version of amyloidosis, amyloid accumulates diffusely and causes myocardial atrophy. In addition, it accumulates in the coronary arteries and aorta.
All of the above changes in the myocardium cause disruption of the systolic and diastolic function of the heart. The patient's hemodynamics are impaired due to a decrease in cardiac output and heart failure develops.
When amyloid damages the tissues of the heart valves, a valve defect is formed. Such conditions are manifested by corresponding symptoms and also lead to hemodynamic disturbances.
If a modified protein is deposited in the conduction system of the heart (bundle of His, sinus or atrioventricular node), then the patient develops signs of arrhythmias.
Stages of flow
Depending on the degree of involvement of organ tissues in the pathological process, four stages of cardiac amyloidosis are distinguished:
- I – with non-invasive methods of examining the heart or biopsy, signs of amyloid accumulation are not detected, the pathology is asymptomatic;
- II – non-invasive methods of cardiac examination or biopsy reveal significant accumulations of amyloid, but the pathology is asymptomatic;
- III – pathology manifests itself with symptoms of cardiomyopathy, but the patient’s condition remains compensated;
- IV – the patient develops decompensated cardiomyopathy, and amyloidosis becomes irreversible.
Diagnosis of cardiac amyloidosis
Definitive diagnosis of amyloidosis requires a biopsy . The abdominal fat layer is often selected for the procedure, since this area is characterized by easy accessibility and low morbidity. However, this area of the body has relatively low sensitivity, so even in positive cases, inadequate amyloid deposits are often obtained, characteristic of the final subtype of the disease (ATTR, AL, etc.). Thus, a biopsy of the organ involved in the pathological process (i.e., the heart if cardiac amyloidosis is suspected) is practiced.
An endomyocardial biopsy provides an almost 100% reliable result about the presence or absence of cardiac amyloidosis.
Determining the subtype of cardiac amyloidosis can be done by immunofluorescence or samples are sent to a laboratory for mass spectrometry. If there is any doubt about the diagnosis based on immunofluorescence staining, mass spectrometry should be performed.
Additional laboratory tests for AL amyloidosis can determine other organ dysfunction:
- proteinuria;
- alkaline phosphatase concentration;
- number of circulating light chains.
Light chain concentration tests may be useful as a presumptive diagnosis before biopsy is performed. In particular, the normal magnitude of circulating light chains makes the diagnosis of AL amyloidosis unlikely. So such studies are necessary to monitor the response to chemotherapy.
For ATTR amyloidosis, genetic testing is performed to determine the transthyretin gene. The presence of a pathological mutation may influence clinical trial options, predict organ involvement, and indicate presumptive family involvement.
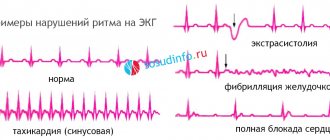
Electrocardiography is an important test for any form of cardiac amyloidosis. With its help, ventricular hypertrophy is detected. Hypertrophy in amyloidosis is the deposition of amyloid fibrils rather than myocyte hypertrophy/hyperplasia. This explains the reduced ECG voltage, rather than increased, as is the case with typical hypertrophy.
Cardiac magnetic resonance imaging is also performed if amyloidosis is suspected. In particular, the global transmural or subendocardial distribution of cardiac tissue can be determined.
Echocardiography - deformities and basal-, apically-predominant disorders are often determined.
Video: Heart with amyloidosis
Symptoms
Amyloid is deposited not only in the heart, but also in many other organs, causing disruption of their function.
For a long time, cardiac amyloidosis is asymptomatic. After this, the patient may experience the following nonspecific symptoms:
- decreased tolerance to physical activity;
- weakness;
- sudden weight loss;
- frequent episodes of dizziness;
- irritability;
- swelling.
All these signs are usually attributed to fatigue and do not become a reason to consult a cardiologist.
More often, the appearance of the first signs of cardiac amyloidosis is provoked by the following factors:
- emotional stress or frequent and prolonged stressful situations;
- acute viral infection.
After exposure to these causes, the patient develops symptoms indicating amyloid damage to heart tissue:
- angina-like chest pain;
- arrhythmic pulse;
- reduction in blood pressure by 20-25% below normal;
- severe weakness;
- presyncope and fainting;
- visual increase in volume of the neck veins;
- dry cough at night (signs of suffocation).
These manifestations of cardiac amyloidosis can be mistaken for other diseases - hypertrophic cardiomyopathy, coronary heart disease. Making a correct diagnosis in such cases is possible only after a comprehensive examination using instrumental and laboratory methods (Echo-CG, myocardial scintigraphy, blood serum analysis, heart biopsy, etc.).
Usually the patient’s condition quickly deteriorates and all the above symptoms can no longer be eliminated by taking cardiac glycosides. These are joined by the following manifestations of progressive heart failure:
- increasing shortness of breath;
- increase in liver size;
- swelling:
- exudative pericarditis;
- ascites.
If the sinus node is damaged, the patient develops bradycardia, which can provoke cardiac arrest and sudden coronary death.
If the accumulation of amyloid in the heart is caused by systemic amyloidosis, then patients have signs indicating damage to other organs and systems:
- increase in tongue size;
- swallowing disorder;
- diarrhea;
- stomach ache;
- intestinal obstruction;
- renal failure;
- itchy skin;
- jaundice;
- dark urine and discoloration of stool;
- periorbital purpura;
- bleeding of mucous membranes;
- hemorrhagic skin rashes;
- severe pain in the shoulder joint.
The variability of the above-described symptoms of generalized amyloidosis can be different and depends on the presence of lesions of certain organs. A patient with this form of the disease develops more and more new symptoms over time.
Diagnostics
Detecting cardiac amyloidosis is difficult because the disease is not accompanied by specific symptoms. A doctor may suspect the presence of this pathology based on the following signs:
- when listening to heart sounds, their dullness and systolic murmur are determined;
- An enlarged shadow of the heart is noticeable on a chest x-ray (there may be signs of fluid accumulation in the heart sac or pleural cavity);
- the ECG reveals disruptions in the excitation of the heart and large-scale deviations in the conduction and excitation systems of the myocardium;
- Echo-CG reveals thickening of the interchamber septa, myocardium, valves and walls of the ventricles, hypokinesia of the heart muscle, dilatation of the atria, and a decrease in the volume of ejection from the left ventricle.
Sometimes, when performing an ultrasound of the heart, it is possible to detect the presence of amyloid deposits in the myocardium in the form of small granules.
To make an accurate diagnosis, the patient is prescribed the following studies:
- biopsy and histological analysis of heart tissue to determine the presence of amyloid;
- blood and urine tests - to detect proteinuria, hypoproteinemia, decreased levels of globulins and albumins.
A more detailed picture of structural changes in the heart due to amyloid damage is provided by the following studies:
- MRI of the heart with contrast;
- myocardial scintigraphy.
To exclude an erroneous diagnosis, differential diagnosis is carried out with the following diseases:
- idiopathic restrictive cardiomyopathy;
- myxedema cardiomyopathy;
- hypertrophic cardiomyopathy;
- Fabry disease;
- aortic stenosis;
- glycogenosis;
- hemochromatosis;
- sarcoidosis;
- pulmonary heart.
The first signs of pathology development
Primary cardiac amyloidosis is asymptomatic, but the presence of a concomitant disease triggers the progression of symptoms.
The first signs indicating the formation of a pathological process are:
- increasing weakness;
- constant fatigue;
- swelling of the lower extremities;
- dyspnea;
- dizziness;
- short-term loss of consciousness;
- interruptions in heart function.
A characteristic sign of the formation of pathology is low blood pressure with a reduced pulse rate
During exercise, the heart is unable to increase cardiac output due to ventricular diastolic dysfunction, atrial damage, or impaired circulation due to vascular amyloid deposition.
A short-term loss of consciousness occurs after suffering emotional or mental stress. This sign indicates an unfavorable course of the disease, since most of these patients expect death.
Treatment
Treatment is predominantly medicinal.
Aimed at improving the functioning of the heart and slowing down the accumulation of amyloid in its cells. So far, modern medicine has not developed effective drugs to completely get rid of amyloidosis. The complex of treatment for this disease is aimed at slowing down the process of amyloid accumulation in tissues and symptomatic therapy. After amyloidosis is detected, the patient should be under medical supervision by a cardiologist, hematologist, nephrologist, neurologist and other specialized specialists.
Conservative treatment
For cardiac amyloidosis, a drug therapy plan is drawn up depending on the root cause of the disease:
- With primary amyloidosis. Cytostatic drugs are prescribed to target plasma cell tumors. The following drugs can be used for this: Melphalan with Prednisolone or Dexamethasone, Thalidomide alone or with chemotherapeutic agents, a combination of Adriamycin, Vincristine with Dexamethasone. Such drug therapy is prescribed in courses and is carried out under the constant supervision of a hematologist and cardiologist. Its side effects are identical to those of chemotherapy and manifest themselves in the form of intoxication, nausea, vomiting, weight loss, and baldness. High doses of Melphalan can provoke the development of leukemia.
- With secondary amyloidosis. Corticosteroids and Colchicine are prescribed. Hormonal drugs have an anti-inflammatory effect, and taking Colchicine helps block the formation of amyloid fractions, is effective in 95% of cases and can increase the patient’s life expectancy.
Research is currently underway on the use of an antitumor drug such as Rituximab, consisting of monoclonal antibodies against the CD20 antigen, for the treatment of cardiac amyloidosis. So far there are no conclusions about the effectiveness of this drug for this pathology.
In addition to these medications, the patient is prescribed symptomatic therapy aimed at eliminating cardiac dysfunction. For this the following can be used:
- anticoagulants;
- diuretics;
- vasodilators;
- Digoxin (with caution);
- vitamins.
When prescribing medications, the doctor uses low dosages, because Taking some of them for amyloidosis can cause an even greater decrease in blood pressure.
If amyloid affects other organs, the treatment plan may include other drugs for symptomatic therapy.
Patients with cardiac amyloidosis are recommended to follow a diet that helps relieve the load on this organ. It involves limiting the amount of salt and animal fats.
In addition to taking medications, patients with severe amyloidosis (especially those with amyloid damage to the kidneys) may be recommended to undergo hemodialysis. This method eliminates the consequences of renal failure and helps prolong the life of patients.
Surgery
In some cases, patients with cardiac amyloidosis may be prescribed the following operations:
- Pacemaker implantation. These interventions are performed when it is impossible to stabilize heartbeats and eliminate arrhythmias with the help of medications.
- Stem cell transplantation. It is carried out using high doses of cytostatics that destroy the bone marrow. The material for transplantation can be taken from the patient himself or from a donor.
- Heart transplantation. Such interventions can be performed in combination with chemotherapy and stem cell transplantation for primary amyloidosis. However, subsequently the donor organ may again be affected by amyloid, and such interventions are carried out only to prolong the patient’s life.
- Transplantation of other organs. For systemic amyloidosis, kidney and liver transplants may be performed.
Description of amyloidosis
The term “amyloidosis” does not refer to one pathology, but to a collection of diseases in which a protein-based infiltrate is deposited in the tissues as beta-pleated sheets. The subtype of the disease is determined by which protein is precipitated, although dozens of subtypes have been described, most of them are incredibly rare or of trivial significance.
Designations
Correct nomenclature uses the letter “A” for amyloid, followed by the letter(s) referring to the major deposited protein. For example, light chain amyloidosis is “AL” (“A” for amyloid and “L” for light chain). Transthyretin amyloidosis is defined as “ATTR” (“A” for amyloid and “TTR” for transthyretin). Terms such as “primary amyloidosis,” “secondary amyloidosis,” “senile amyloidosis,” and “familial amyloid cardiomyopathy” are often confusing and are best avoided.
The vast majority of cardiac amyloidosis is caused by one of two proteins: light chains or transthyretin. The clinical manifestations and treatment of these two subtypes differ.
Light chain amyloidosis (AL amyloidosis)
It is the most commonly diagnosed form of systemic amyloidosis. Also formerly defined as “primary amyloidosis”, but for the reasons given above this name will not be used.
Plasma cells are found primarily in the bone marrow and produce large amounts of antibodies. Antibodies are composed of heavy chains and light chains. When a plasma cell becomes clonal (essentially malignant), it and its clones typically produce a clonal antibody and a clonal light chain excess associated with that antibody. There are three possible outcomes in this process:
- The plasma cell clone takes over a small portion of the bone marrow, and the light chain is excreted harmlessly in the urine. This condition is called monoclonal gammopathy of undetermined significance.
- The plasma cell clone engulfs most of the bone marrow, potentially leading to hypercalcemia, anemia, lytic lesions, and/or renal dysfunction. This condition is called myeloma.
- The plasma cell clone produces a light chain that is prone to being incorrectly replaced by beta-pleated sheets (β-pleated sheet). These light chains circulate in the bloodstream and are deposited in one or more tissues. This condition is called AL amyloidosis.
AL amyloid deposits can occur in virtually any tissue, and the pattern of organ involvement varies from patient to patient (Table 1).
Common extracardiac sites of involvement and associated manifestations are:
- kidneys (albuminuria and potential renal failure);
- liver (increased alkaline phosphatase and potential liver failure);
- gastrointestinal tract (dysphagia, constipation, malabsorption and bleeding);
- tongue (macroglossia);
- nervous system (peripheral neuropathy and autonomic dysfunction).
In extremely rare cases, clinical involvement of all these organs and systems is determined in one patient.
Table 1: Amyloid subtypes and clinical characteristics
| Subtype | Demography | Organ involvement | Left ventricular hypertrophy | Therapy |
| AL | M ≈ F Age 40-80 | Any (heart, kidney, GM, tongue, nerves, liver, soft tissues) | + | Chemotherapy or stem cell transplant |
| Wild type ATTR M | >>> F Age 65-95 | Heart (carpal tunnel syndrome) | +++ | Maintenance therapy, clinical trials ongoing |
| Mutant ATTR | M >> F Age 55-75 | Heart and nerves (carpal tunnel syndrome) | +++ | Maintenance therapy, clinical trials ongoing |
Cardiac manifestations of amyloidosis are as follows:
- Heart failure (HF), which can be diastolic or systolic.
- Arrhythmias (tachyarrhythmias/bradyarrhythmias).
The main feature is ventricular hypertrophy , seen on very low voltage electrocardiogram (ECG) echocardiography.
Natriuretic peptides are typically elevated in cardiac amyloidosis, and troponin assays are often chronically positive at low levels (0.1–1 ng/mL) due to the ongoing gradual destruction of cardiomyocytes.
Transthyretin amyloidosis (ATTR amyloidosis)
Transthyretin (prealbumin) is a protein produced by the liver that functions as a transporter for thyroxine and retinol. It predominantly circulates as a homotetramer with a small amount of transthyretin present in monomeric form. This form of transthyretin is subject to improper replacement and is gradually deposited as amyloid deposits.
There are two main subtypes of ATTR amyloidosis:
- ATTR wild type
- Mutant ATTR.
In the case of wild-type ATTR, the transthyretin protein is normal (unmutated). Over decades, it is gradually deposited as amyloid accumulations. Although small amounts of deposits may occur in the soft tissue (causing carpal tunnel syndrome) and vasculature, the primary pathological deposits are found in the heart.
ATTR mutant cardiac amyloidosis is an inherited disease. It detects pathological mutations in the transthyretin gene, which leads to accelerated amyloid deposition. Mutant ATTR most often affects nerve fibers, and the pattern of deposition largely depends on the mutation.
Forecast
The prognosis for cardiac amyloidosis is always unfavorable due to rapidly progressing heart failure. This pathology can be complicated by myocardial infarction, ischemic stroke and the development of infectious processes in various systems and organs (pneumonia, glomerulonephritis, erysipelas, etc.). The most dangerous complication of this disease is sudden coronary death.
The onset of death with cardiac amyloidosis occurs approximately 1.5 - 2.5 years after detection of the pathology. The cause of death can be various factors - heart failure, cardiac or non-cardiac complications of amyloidosis.
Cardiac amyloidosis is a rare but dangerous disease. Due to the accumulation of amyloid in the tissues of the heart, cardiac function is impaired. This process is irreversible, and treatment for such a disease can only be aimed at prolonging the patient’s life.
Channel One, program “Live Healthy!” with Elena Malysheva, in the “About Medicine” section, a conversation about amyloidosis: