Etiology
Thalassemia is caused by point mutations or deletions in the genes encoding hemoglobin chains. As a result, this can lead to a decrease in synthesis or complete absence of one of the chains in the body. The other chain forms inadequate hemoglobin tetramers, which leads to the destruction of red blood cells and hemolytic anemia.
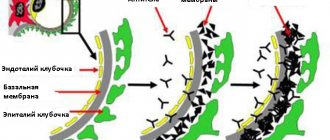
The structure of hemoglobin
Hemoglobin is a protein found in red blood cells that is responsible for transporting oxygen to tissues and carbon dioxide from them.
Hemoglobin (we are talking about HbA) consists of four chains: two alpha subunits and two beta subunits. This hemoglobin makes up 97% of the total content in red blood cells.
The remaining 3% is hemoglobin HbA2, which differs in the structure of its two chains: instead of beta subunits, it has delta subunits. HbA and HbA2 are normal when they are in the correct ratio.
Each of the hemoglobin chains binds to its non-protein part - heme.
So, with thalassemia, the synthesis of one of the hemoglobin chains is disrupted: either alpha or beta. According to this principle, there is a classification of thalassemia into:
- alpha thalassemia;
- beta thalassemia.
Thalassemia is classified according to severity:
- mild;
- moderate;
- severe.
Types of hemoglobin
There are physiological and pathological types of hemoglobin.
Hemoglobin, which may be normal in humans, includes:
- HbP – primitive hemoglobin, found in the embryo between the 7th and 12th weeks of life;
- HbF – fetal hemoglobin, contains two alpha and two gamma chains, appears after 12 weeks of intrauterine development, in adults its content is less than 1%;
- HbA – hemoglobin of adults, the proportion is 97%, contains two alpha and two beta chains;
- HbA2 – hemoglobin of adults, the proportion is 2%, contains two alpha and two delta chains,
- HbO2 – oxyhemoglobin, formed when oxygen binds in the lungs;
- HbCO2 – carbohemoglobin, is formed when carbon dioxide binds in tissues.
Pathological forms of hemoglobin include:
- HbS – hemoglobin, determined in sickle cell anemia;
- MetHb – methemoglobin, contains a trivalent iron ion, when normally it is divalent. This form is formed when consuming sulfonamides, nitrates, or vitamin C deficiency. Methemoglobin is not able to bind oxygen, resulting in tissue hypoxia;
- HbCO – carboxyhemoglobin, is formed when there is excess carbon monoxide in the inhaled air. It is present in small concentrations in the blood, but its level may increase depending on the characteristics of the inhaled air.
HbS is a type of hemoglobin that occurs when the beta chain is mutated (one amino acid is replaced by another). It occurs in people with sickle cell anemia. Red blood cells containing such hemoglobin do not live long and are quickly destroyed, which is good in the habitats of the malarial plasmodium. People with sickle cell disease have resistance to this parasite.
Frequency of occurrence
Thalassemia is inherited in an autosomal recessive manner. This means that it affects both boys and girls equally. The recessive nature suggests that children with thalassemia appear in those families where mom and dad are both carriers of mutations. Although they may not even realize that they are carriers, since the symptoms are sometimes invisible.
The average incidence is 1 in 100,000 people and may vary by region. Beta thalassemia is more common than alpha thalassemia.
What to do if a child is diagnosed with thalassemia minor?
As I already said, with this form of the disease there are almost no external symptoms. At an early age, the child will experience a slight decrease in hemoglobin, which, as a rule, does not require treatment. His erythrocyte indices will remain reduced for the rest of his life, which is not dangerous to health. Otherwise, such a child, and then an adult, is no different from other people . He can be considered a carrier of the thalassemia gene rather than a sick person. In women who carry this gene, mild anemia may return during pregnancy.
Important!
Alpha thalassemia
There are 4 gene regions that encode the synthesis of the alpha chain of hemoglobin. If there is a genetic defect (mutation), alpha thalassemia occurs in them. There are several forms of alpha thalassemia.
Homozygous
When it occurs, the synthesis of alpha subunits in all four sections of the gene is disrupted. As a result, hemoglobin is formed, consisting entirely of beta chains. It is also called Barth's hemoglobin. Such hemoglobin has a high affinity for oxygen. This means that it has difficulty delivering oxygen to tissues. As a result, a lack of this gas occurs, which leads to the development of heart failure, edema, dropsy and, as a consequence, intrauterine fetal death.
H-hemoglobinopathy
H-hemoglobinopathy is a form in which the synthesis of the alpha chain in three gene regions is disrupted. As a result of excess beta subunits, HbH is formed. It also has a fairly high affinity for oxygen, it is not stable, and is easily oxidized.
The presence of such hemoglobin in red blood cells changes the red blood cell membrane and is accompanied by the development of hemolytic anemia, that is, increased breakdown of red blood cells. This form of alpha thalassemia appears by the age of 1 year in the form of hemolytic anemia. There are signs of anemic syndrome and an enlarged spleen. Blood tests show increased reticulocyte values, which indicates an increased regenerative ability of the bone marrow to restore damaged cells. The red blood cells are hypochromic and target-like.
Alpha thalassemia minor
Alpha thalassemia minor is a form in which the synthesis of 1 or 2 alpha chains is impaired. With this form there are no severe clinical manifestations. A mild degree of anemia (microcytic, hypochromic) may be observed in the blood.
Despite the fact that 1 or 2 genes responsible for the synthesis of alpha subunits are disrupted, this does not lead to such serious disorders as, for example, with beta thalassemia.
Symptoms of thalassemia in children
Based on clinical manifestations, doctors divide the symptoms of the disease into three types: major, minor, minimal. There are also transitional types between these types.
The large type, homozygous carriage of the disease (Cooley's disease), is manifested by the following clinical signs:
- hemolytic type anemia;
- hepatosplenomegaly;
- osteoporosis of the flat bones of the skull.
Small type, heterozygous carriage is manifested by clinical signs:
- anemia of various origins;
- splenomegaly, hepatosplenomegaly (liver size is increased less than in large types);
- changes in the structure of the flat bones of the skull.
The minimal type of thalassemia is asymptomatic. Clinically manifested in parasitic infections, pregnancy, injuries with large blood loss.
Beta thalassemia
Beta thalassemia occurs when the synthesis of beta subunits is impaired. Due to mutations, hemoglobin is formed in which the beta chains are replaced by alpha. Alpha tetramers are formed in red blood cells. The reticuloendothelial system removes them from the red blood cells, they are damaged, and hemolysis occurs.
As a result of mutations in the beta chain gene, beta thalassemia major and beta thalassemia minor are distinguished.
Beta thalassemia minor
Beta thalassemia minor (minor) is a heterozygous form characterized by a mild course.
In laboratory tests:
- the number of sideroblasts is normal or increased;
- hypochromic microcytic anemia;
- anisocytosis - red blood cells of different sizes;
- poikilocytosis - red blood cells of different shapes;
- increased number of reticulocytes;
- iron is normal;
- elevated levels of indirect bilirubin.
In patients with beta thalassemia minor, there is an increase in the level of HbA2 (up to 6%, normal is up to 3%) and HbF (up to 7%, normal is less than 1%).
Beta thalassemia minor is often confused with iron deficiency anemia and is mistakenly treated with iron supplements. To differentiate between these two conditions, it is necessary to evaluate the level of iron in the blood. With beta thalassemia it will be within normal limits.
Thalassemia major
Thalassemia major (Cooley's anemia) is homozygous and is considered a severe progressive form of beta thalassemia. Like other forms of thalassemia, it is manifested by pale skin and an enlarged spleen. They appear by the child's 1st year of age. A feature of Cooley's anemia is bone changes: protruding cheekbones, narrow palpebral fissures, square skull, flat bridge of the nose. Such children have low physical development.
Laboratory tests indicate:
- increased content of sideroblasts;
- low MCV, MCH, MSHC values, indicating hypochromic microcytic anemia;
- anisocytosis;
- poikilocytosis – in the form of a target, schizocytes;
- basophilic puncture of erythrocytes;
- increased osmotic resistance of red blood cells;
- increase in unconjugated bilirubin;
- excess iron content, up to its deposition in organs (hemosiderosis).
Beta thalassemia major is characterized by an increase in the content of fetal (HbF) hemoglobin in the blood to 70%, which is confirmed by electrophoresis.
Sideroblasts - they are also called erythroblasts. They contain non-hemoglobin iron in the cytoplasm in the form of hemosiderin and ferritin.
Possible reasons
As we have already found out, thalassemia is caused by inheritance of a mutant gene in an autosomal recessive manner . Therefore, children who have both parents with a predisposition to thalassemia can get sick. In other words, if they received the corresponding gene from both their mother and father. If the anomaly is observed in one of them, the likelihood of getting sick is reduced (from 25% to 50%), and clinical manifestations will be either mild or completely absent.
Autosomal recessive mode of inheritance
But the main problem with thalassemia (like many other hereditary pathologies) is that a healthy genome can mutate. Or, to put it another way, there is still a chance of “getting sick” in this case, although it is quite small. What factors can provoke a mutation?
- Ionizing radiation and increased background radiation. To do this, it is not at all necessary to live near a nuclear reactor: it is enough, for example, to use a mobile phone too actively or sit for many hours near a computer monitor. And Chernobyl, although the accident happened almost 30 years ago, will remind itself for a long time. This effect can also be achieved by enhanced radiotherapy for cancer or repeated X-ray examinations (especially with outdated equipment).
Modern equipment is much safer for health
- Various chemicals with a strong mutagenic effect. Those at risk are those working in hazardous industries, although one should not discount an excessive passion for low-quality food products.
- Pharmacological drugs. Strong drugs can also provoke a mutation. These are mainly cytostatics (drugs aimed at combating malignant tumors) of previous generations.
- Viruses: herpes, measles or even regular flu.
- Smoking. The main culprits are harmful chemical compounds contained in tobacco smoke (phenol, benzene).
- Low-quality cheap alcohol or abuse of strong drinks.
Common clinical signs of thalassemia
- Hemolytic anemia will lead to pallor, lethargy and sometimes even yellowing of the skin .
- The abdomen will be enlarged due to splenomegaly (entrainment of the spleen).
- In some forms of thalassemia, bone deformities .
- Children with thalassemia experience delayed physical development due to a lack of oxygen for tissue growth and development.
- Due to increased absorption of iron in the intestines and frequent blood transfusions, an increase in this element in the blood is observed; it is deposited in organs and tissues (for example, heart, liver), disrupting their functioning.
Signs and symptoms of the disease
Signs of thalassemia in children have vivid symptoms, the degree of its expressiveness depends on the complexity of the disease and appears in all its colors closer to one year of age.
Representative external signs of the presence of the disease are:
- Changes in skull morphology. The head of a child with a complex degree of thalassemia acquires an unnatural structure in the form of a square, the upper jaw is characterized by enlarged dimensions. The bridge of the nose has a smoothed shape, and the face acquires Mongoloid features.
- Pale skin color. The color of the skin can vary from gray to yellow in unnatural shades.
- Signs of physical and psychological retardation are expressed.
- Enlargement of the spleen and liver, which can be detected by palpation, without special studies.
Symptoms of thalassemia have signs similar to anemic indicators:
- Headache and frequent dizziness, often accompanied by loss of consciousness.
- Loss of strength and decreased capacity for no obvious reason.
- The occurrence of shortness of breath without heavy physical activity.
Thalassemia is characterized by the presence of excess iron in the body, which negatively affects the functionality of the cardiac system and may result in arrhythmia or heart failure. Symptoms of the accumulation of excess iron in the liver can include a decrease in the patient’s appetite, a decrease in his body weight, the presence of frequent infectious diseases, as well as a decrease in blood clotting. Deposits of iron components in the kidneys and lungs can manifest as renal failure and pathologies of the respiratory functions of the body. Gastritis and joint diseases are frequent companions of the disease; their fact is explained by the consequence of hemosiderosis, characteristic of the disease.
The most popular drug used in the treatment of thalassemia is Desferal
Diagnosis of thalassemia
When diagnosing thalassemia, one cannot rely only on the clinic alone, especially since it may not always be present, for example, in mild forms. Therefore, for an accurate diagnosis it is necessary to “look” inside the body and evaluate the processes occurring in it. Laboratory blood tests will help with this.
Anemia will be indicated by low levels of hemoglobin and red blood cells . A microscopic examination of the blood smear will reveal the shape of the red blood cells and the hemoglobin content in them. That is, the microcytic hypochromic nature of anemia will be confirmed.
Hemoglobin electrophoresis will allow you to assess how its fractional composition has changed (the ratio of HbA, HbA2, HbF to each other).
An ultrasound examination will show an increase in the size of the spleen.
X-rays are used to study bone deformities.
Bone marrow puncture will allow you to evaluate the processes of hematopoiesis and make a conclusion about the severity of the processes.
The main study that can confirm the presence of thalassemia is molecular genetic analysis . An identified mutation in the alpha or beta chain gene indicates thalassemia.
A biochemical blood test will determine a number of indicators that may increase with thalassemia, for example, indirect bilirubin. Assessing iron metabolism in the body will allow for differential diagnosis between iron deficiency anemia and thalassemia.
Before starting treatment, it is necessary to conduct a genetic study to determine the type of mutation and further prognosis for the development of the disease.
Prevention
Prevention of the disease includes:
- Prenatal diagnosis. Persons suffering from the disease should be examined by a geneticist during pregnancy planning.
- Fetoscopy and amniocentesis. These procedures are performed when both parents have thalassemia. Using a puncture, fetal tissue samples are obtained and undergo genetic testing. The procedure is performed under ultrasound guidance. The methods have side effects, they can provoke spontaneous abortion, intrauterine infection or premature birth. If positive test results are received, the woman is recommended to terminate the pregnancy.
Treatment of thalassemia
Thalassemia is a hereditary disease, so there is only symptomatic therapy.
Treatment for thalassemia will depend on the form. For mild forms, therapy is not carried out. If necessary, blood transfusions . Patients with beta thalassemia need to monitor their blood iron levels and, if they have excess levels, they should be prescribed iron-binding medications .
Patients with severe forms are prescribed blood transfusions in combination with drugs that remove iron from the body. Hemoglobin is maintained at 100 g/l.
Deferoxamine is one of those drugs that can form complexes with iron and remove it in the urine. Do not take anything on your own, only take all medications as prescribed by your doctor.
If symptoms do not improve after a blood transfusion, surgery may be performed to remove the spleen.
What complications can there be?
Thalassemia can lead to the development of other health problems:
- Enlarged spleen.
The spleen helps the body fight infections and filter damaged blood cells. With thalassemia, the work of the spleen may become more intense, causing it to increase in size. If the enlargement is critical, the spleen often needs to be removed. - Infections.
People with thalassemia are more likely to get blood infections, especially if they receive frequent transfusions. Some types of infections are made worse by removal of the spleen. - Bone problems.
Thalassemia can lead to bone deformities of the face and skull. People with this disease also often develop severe osteoporosis (brittle bones). - Excess iron in the blood.
This disruption can in turn cause damage to the heart, liver, and endocrine system (such as the thyroid and adrenal glands).
What should I do if I am a carrier of thalassemia and want to get pregnant?
Some particularly severe types of thalassemia cause the death of the baby before or shortly after birth. If you or your partner know that you are a carrier of thalassemia, you should definitely consult with a specialist or genetic counselor before conceiving. Certain tests can show which type of disease you are carrying. Once you become pregnant, you can find out whether your fetus has thalassemia through prenatal testing.
(No Ratings Yet)
Loading…